تنزيل العرض التّقديمي
العرض التّقديمي يتمّ تحميله. الرّجاء الانتظار

1
كلية العلوم للبنات - جامعة الملك عبد العزيز أستاذ الكيمياء الفيزيائية
كلية العلوم للبنات - جامعة الملك عبد العزيز قسم الكيمياء الكيمياء الفيزيائية (حركية ) Kinetics - Chemistry رمز المقرر : CHME رقم المقرر : 344 الفصل الدراسي الثاني : 1432هـ إعداد/ أ .د .سناء طاهر عرب أستاذ الكيمياء الفيزيائية
 Kinetics - Chemistry. رمز المقرر : CHME. رقم المقرر : 344. الفصل الدراسي الثاني : 1432هـ. إعداد/ أ .د .سناء طاهر عرب. أستاذ الكيمياء الفيزيائية.")
2
الكيمياء الحركية

3
الكيمياء الحركية هو العلم الذي يختص بدراسة سرعة التفاعلات الكيميائية والعوامل المؤثرة فيها

4
معدل سرعة التفاعل معدل التفاعل أو معدل سرعة التفاعل الكيميائي هو معدل التغير في كميات (تركيز) المواد المتفاعلة أو الناتجة في وحدة الزمن. أو معدل تغير نواتج التفاعل في وحدة الزمن أي أنه عبارة عن الزيادة في تركيز المواد الناتجة بمرور الزمن وتكوين الناتج.
 المواد المتفاعلة أو الناتجة في وحدة الزمن. أو معدل تغير نواتج التفاعل في وحدة الزمن أي أنه عبارة عن الزيادة في تركيز المواد الناتجة بمرور الزمن وتكوين الناتج.")
5
معدل سرعة التفاعل وباعتبار قانون بقاء المادة فإن زيادة تركيز النواتج يعني نقص تركيز المتفاعلات. وعملياً يمكن قياس سرعة التفاعل بقياس معدل اختفاء مادة متفاعلة أو معدل تكوين مادة ناتجة. وعادةً يتم ذلك بمتابعة تغير احدى الخواص الفيزيائية للمادة.

6
A B time

7
سرعة التفاعل الكيميائي:
تحتاج أي ظاهرة كيميائية أو فيزيائية إلى وقت ما لتحدث قد يكون هذا الزمن قصيراً جداً وقد يكون طويلاً جداً. وكثير من التفاعلات المألوفة والتي يتم إجراؤها في المختبر في أنبوبة اختبار هي تفاعلات سريعة جداً فإذا أضيف كلوريد الباريوم إلى محلول كبريتات الصوديوم يلاحظ تكون راسباً أبيضاً في أنبوبة الاختبار هو عبارة عن كبريتات الباريوم حسب المعادلة: Ba Cl2 + Na2 SO Ba SO Na Cl وأيضاً تفاعلات التعادل بين محاليل الأحماض والقلويات تحدث في وقت لا يمكن قياسه لقصره حيث يتحد أيون الهيدروكسيل مع أيون الهيدروجين ويتكون الماء OH- + H H2O ويستدل على ذلك من سرعة تغير لون الدليل المستخدم في الدراسة . بينما تفاعل غاز الأكسجين مع غاز الهيدروجين لتكوين الماء لا يحصل في الأحوال العادية حتى ولو بقيا في وعاء واحد أياما عديدة. O2 + 2 H H2O وهناك أمثلة عديدة لتفاعلات كيميائية أخرى بطيئة جداً تأخذ أزمنة طويلة مثل تفاعلات التربية الجيولوجية وتكون الصدأ على الحديد. سرعة التفاعل الكيميائي: تحتاج أي ظاهرة كيميائية أو فيزيائية إلى وقت ما لتحدث قد يكون هذا الزمن قصيراً جداً وقد يكون طويلاً جداً. وكثير من التفاعلات المألوفة والتي يتم إجراؤها في المختبر في أنبوبة اختبار هي تفاعلات سريعة جداً فإذا أضيف كلوريد الباريوم إلى محلول كبريتات الصوديوم يلاحظ تكون راسباً أبيضاً في أنبوبة الاختبار هو عبارة عن كبريتات الباريوم حسب المعادلة: Ba Cl2 + Na2 SO Ba SO Na Cl وأيضاً تفاعلات التعادل بين محاليل الأحماض والقلويات تحدث في وقت لا يمكن قياسه لقصره حيث يتحد أيون الهيدروكسيل مع أيون الهيدروجين ويتكون الماء OH- + H H2O ويستدل على ذلك من سرعة تغير لون الدليل المستخدم في الدراسة . بينما تفاعل غاز الأكسجين مع غاز الهيدروجين لتكوين الماء لا يحصل في الأحوال العادية حتى ولو بقيا في وعاء واحد أياما عديدة. O2 + 2 H H2O وهناك أمثلة عديدة لتفاعلات كيميائية أخرى بطيئة جداً تأخذ أزمنة طويلة مثل تفاعلات التربية الجيولوجية وتكون الصدأ على الحديد.

8
سرعة التفاعلات وقوانينها
جهاز طيف الإمتصاص

9
(rate r )سرعة التفاعل
سرعة التفاعل")
10
CH4 (g) + 2O2 (g) CO2 (g) + 2H2O (g)
rate = - d[CH4] dt = - d[O2] dt 1 2 = d[CO2] dt = d[H2O] dt 1 2 2H2 + O H2O وحدة سرعة التفاعل (Concentration) (time)-1 والوحدة الشائعة هي : mol l-1s-1
 (time)-1. والوحدة الشائعة هي : mol l-1s-1.")
11
-3.5 x10-2 مول لكل لتر و لكل ثانية فاحسب :
مثال : عندما تتفاعل الامونيا (غاز النشادر ) مع الأكسجين عند درجات الحرارة العالية يتكون اكسيد النيتروجين (NO) وبخار الماء فاذا علمت ان سرعة اختفاء الأمونيا تساوى -3.5 x10-2 مول لكل لتر و لكل ثانية فاحسب : ا) سرعة اختفاء الأكسجين ب) سرعة تكوين اكسيد النيتروجين ج) سرعة تكوين الماء 4NH3(g)+5O2(g) → 4NO(g)+6H2O(g) سرعة اختفاء الأكسجين تساوى 1.25سرعة اختفاء الأمونيا
 مع الأكسجين عند درجات الحرارة العالية. يتكون اكسيد النيتروجين (NO) وبخار الماء فاذا علمت ان سرعة اختفاء الأمونيا تساوى x10-2 مول لكل لتر و لكل ثانية فاحسب : ا) سرعة اختفاء الأكسجين ب) سرعة تكوين اكسيد النيتروجين ج) سرعة تكوين الماء. 4NH3(g)+5O2(g) → 4NO(g)+6H2O(g) سرعة اختفاء الأكسجين تساوى 1.25سرعة اختفاء الأمونيا.")
12
=1. 5x3.5x10-2 = 5.25x Ms-1

13
العوامل المؤثرة علي سرعة التفاعلات
طبيعة المواد المتفاعلة التركيز درجة الحرارة وجود الحافز

14
اختزال أيونات البرمنجنات MnO4- بواسطة أيونات الحديدوز في الوسط الحامضي
1.طبيعة المواد المتفاعلة: تختلف سرعة التفاعل الكيميائي باختلاف المواد المتفاعلة واختلاف الوسط الذي يجرى فيه التفاعل ، وعملياً وجد أن طبيعة المواد المتفاعلة تؤثر تأثيراً واضحاً في سرعة التفاعل ولعل هذا يتضح من المثال التالي: اختزال أيونات البرمنجنات MnO4- بواسطة أيونات الحديدوز في الوسط الحامضي Fe Fe Mn MnO4 - تتم بسرعة ويلاحظ ذلك من اختفاء لون البرمنجنات مما يدل على إن التفاعل سريع بينما اختزال نفس الأيونات بواسطة حمض الاكساليك في الوسط الحامضي يتم على مهل مما يدل على أن التفاعل بطئ. 2H2C2O H H2O + 4CO Mn MnO4 -

15
2.تركيز المواد المتفاعلة:
لوحظ أنه عندما يكون تركيز المواد المتفاعلة كبيراً وذلك عند بداية التفاعل تكون سرعة التفاعل كبيرة وبمرور الوقت يقل تركيز المواد المتفاعلة وتنخفض معه سرعة التفاعل . عموماً العلاقة بين تركيز المواد المتفاعلة وسرعة التفاعل ليست بالعلاقة السهلة حيث تؤدي الزيادة في تركيز إحدى المواد المتفاعلة إلى زيادة سرعة التفاعل وقد تؤدي إلى نقصانها كما في التفاعلات العكسية وقد لا يحدث أي تغيير في السرعة وأحياناً قد تؤدي إلى تسمم التفاعل ولاسيما في التفاعلات غير المتجانسة.

16
تأثير درجة الحرارة على سرعة التفاعل و طاقة التنشيط
تؤثر درجة الحرارة على سرعة التفاعل تأثيرا كبيرا ووجد عمليا ان رفع درجة الحرارة يؤدى الى زيادة ملحوظة فى سرعة التفاعل ومن ثم من قيمة ثابت سرعة التفاعل. (Arrhenius equation) معادلة ارهينيوس k = A • exp( -Ea/RT ) k ثابت سرعة التفاعل R الثابت العام للغازات (8.314 J/K mol) Ea طاقة التنشيط ووحداتها (J/mol) A معامل التردد Tدرجة الحرارة المطلقة
 معادلة ارهينيوس. k = A • exp( -Ea/RT ) k ثابت سرعة التفاعل. R الثابت العام للغازات (8.314 J/K mol) Ea طاقة التنشيط ووحداتها (J/mol) A معامل التردد. Tدرجة الحرارة المطلقة.")
17
طاقة التنشيط طاقة التنشيط A + B C + D تفاعل ماص للحرارة تفاعل طارد للحرارة طاقة التنشيط : هى أقل مقدار من الطاقة تحتاجه الجزيئات المتفاعلة لكى تصل الى الحالة النشطة ويحدث التفاعل وتكون قادرة على تكوين نواتج

18
طاقة التنشيط

19
تأثير درجة الحرارة على سرعة التفاعل و طاقة التنشيط
معادلة ارهينيوس ln k = ln A - Ea/RT ln k2 k1 = Ea RT - 1 T2 T1

20
حساب طاقة التنشيط بيانيا
ln k = -Ea/R (1/T) + ln A حساب طاقة التنشيط بيانيا
 + ln A. حساب طاقة التنشيط بيانيا.")
21
مثال إذا علمت أن تفاعل الأمونيا مع ثانى اكسيد الكربون فى مراحله الأولية من الرتبة الثانية وكانت قيمة ثابت سرعة التفاعل عند درجة حرارة 327°C تساوى M-1 s-1 وعند درجة حرارة 443°C اصبحت 16.0 M- 1 s-1 فاحسب طاقة التنشيط ب) معامل التردد (ثابت أرهينيوس ) 𝑙𝑛 𝑘 2 𝑘 1 = − 𝐸 𝑎 𝑅 1 𝑇 2 − 1 𝑇 1 ln(16.0/0.385) = Ea/8.314X { }/716x600 Ea = 114x103 J/mol =114.8 kJ/mol ln(16) = ln(A) x103/ 8.314x716 A = 3.8x109 M-1s-1
 معامل التردد (ثابت أرهينيوس ) 𝑙𝑛 𝑘 2 𝑘 1 = − 𝐸 𝑎 𝑅 1 𝑇 2 − 1 𝑇 1. ln(16.0/0.385) = Ea/8.314X { }/716x600. Ea = 114x103 J/mol =114.8 kJ/mol. ln(16) = ln(A) x103/ 8.314x716. A = 3.8x109 M-1s-1.")
22
تأثير درجة الحرارة على سرعة التفاعل و طاقة التنشيط
تمرين: إذا علمت ان طاقة التنشيط لتفاعل ما تساوى 146.3kJ/mol. فاحسب بأى نسبة سوف تزداد قيمة ثابت سرعة التفاعل عندما تزداد درجة الحرارة من °C 𝑙𝑛 𝑘 2 𝑘 1 = − 𝐸 𝑎 𝑅 1 𝑇 2 − 1 𝑇 1

23
وجود الحافز: الحوافز عبارة عن مواد لها خاصية تنشيط التفاعل الكيميائي وزيادة سرعته دون أن تتغير كيميائياً(حفز موجب ) ودورها هو الإسراع في إتمام التفاعل عن طريق مسالك تكون الطاقة التنشيطية اللازمة لها أقل بكثير من الطاقة التنشيطية اللازمة لإتمام التفاعل بدون الحــافز , وقد يقوم عامل الحفز في بعض الأحـــــيان بخفض سرعة التفاعل الكيميائي ويسمى في هذه الحالة مثبط للتفاعل (حفز سالب ). وهناك أنواع كثيرة مختلفة من الحوافز، كما تختلف الطريقة التي يعمل بها ولاسيما من ناحية تكوين المركب المتوسط. وقد يكون الحافز مادة غازية أو سائلة أو صلبة أو قد يكون ذاتياً. ومن دراسة سرعة التفاعل الكيميائي ومعرفة أثر العوامل المذكورة يمكن معرفة الكثير من تفاصيل الخطوات التي يتم بها تحويل المواد المتفاعلة إلى مواد ناتجة.
 ودورها هو الإسراع في إتمام التفاعل عن طريق مسالك تكون الطاقة التنشيطية اللازمة لها أقل بكثير من الطاقة التنشيطية اللازمة لإتمام التفاعل بدون الحــافز , وقد يقوم عامل الحفز في بعض الأحـــــيان بخفض سرعة التفاعل الكيميائي ويسمى في هذه الحالة مثبط للتفاعل (حفز سالب ). وهناك أنواع كثيرة مختلفة من الحوافز، كما تختلف الطريقة التي يعمل بها ولاسيما من ناحية تكوين المركب المتوسط. وقد يكون الحافز مادة غازية أو سائلة أو صلبة أو قد يكون ذاتياً. ومن دراسة سرعة التفاعل الكيميائي ومعرفة أثر العوامل المذكورة يمكن معرفة الكثير من تفاصيل الخطوات التي يتم بها تحويل المواد المتفاعلة إلى مواد ناتجة.")
24
ولما كان وجود الحوافز يضاعف من سرعة التفاعل بمقدار عشرات الأضعاف وقد يكون الحافز سطح مادة صلبة كجدار إناء التفاعل. لذلك يمكن تقسيم التفاعلات الكيميائية من وجهة نظر حركية التفاعلات إلى: تفاعلات متجانسة Homogeneous Reactions تفاعلات غير متجانسة Heterogeneous Reactions والتفاعل المتجانس: هو الذي يحدث كلياً في طور واحد (One Phase) بينما التفاعل غير المتجانس: هو الذي يحدث جزئياً على الأقل في أكثر من طور واحد. وأغلب التفاعلات غير المتجانسة العادية تكون لها سرعة تفاعل تعتمد على مساحة السطح التي يتعرض لها خليط المواد المتفاعلة وهذا السطح يمكن أن يكون الجدار الداخلي لإناء التفاعل أو يمكن أن يكون المادة الصلبة الحافزة. لذلك عند دراسة حركية التفاعل لابد من التأكد من تأثير جدارة الإناء في سرعة التفاعل في أي مرحلة من مراحل التفاعل، ويمكن ذلك في حالة كون جدار الإناء من الزجاج فتضاف كرات زجاجية صغيرة أو أنابيب شعرية رقيقة يحدث على سرعة التفاعل. فإذا كان التفاعل متجانساً فلن تتأثر سرعته، أما إذا كان التفاعل غير متجانس فإن سرعته تتأثر بزيادة السطح المعرض.
 بينما التفاعل غير المتجانس: هو الذي يحدث جزئياً على الأقل في أكثر من طور واحد. وأغلب التفاعلات غير المتجانسة العادية تكون لها سرعة تفاعل تعتمد على مساحة السطح التي يتعرض لها خليط المواد المتفاعلة وهذا السطح يمكن أن يكون الجدار الداخلي لإناء التفاعل أو يمكن أن يكون المادة الصلبة الحافزة. لذلك عند دراسة حركية التفاعل لابد من التأكد من تأثير جدارة الإناء في سرعة التفاعل في أي مرحلة من مراحل التفاعل، ويمكن ذلك في حالة كون جدار الإناء من الزجاج فتضاف كرات زجاجية صغيرة أو أنابيب شعرية رقيقة يحدث على سرعة التفاعل. فإذا كان التفاعل متجانساً فلن تتأثر سرعته، أما إذا كان التفاعل غير متجانس فإن سرعته تتأثر بزيادة السطح المعرض.")
25
رتبة التفاعل n = σ + β n = رتبة التفاعل الكلية
α و β عبارة عن اعداد مجردة ممكن ان تأخذ قيما موجبة أو سالبة أو كسرية أو صفر وتعرف بالرتب الجزئية . n = σ + β n = رتبة التفاعل الكلية

26
(Rate Law) قانون سرعة التفاعل
لا يمكن التنبؤ بشكل عام بقانون سرعة التفاعل بمجرد النظر للمعادلة الكيميائية الموزونة بل يجب معرفة ذلك بالتجربة العملية ويسمى بقانون سرعة التفاعل التجريبي . يقصد بدرجة التفاعل الكيميائي أس المكونات المشتركة في التفاعل والتي يؤثر تركيزها في سرعة التفاعل أو بعبارة أخرى تتحدد درجة التفاعل من مجموع أسس حدود تركيزات المواد الداخلة في الخطوة المحددة لسرعة التفاعل. ويمكن استخدام الطريقة التي تتغير بها سرعة التفاعل مع تركيز المواد المتفاعلة للتعبير عن درجة التفاعل فالتفاعل الذي سرعته تعتمد على الأس الأول لتركيز مادة من المواد المتفاعلة يسمى تفاعلاً من الدرجة الأولى First order reaction بينما يكون التفاعل الذي تتناسب سرعته مع الأس الصحيح لتركيز مادتين من المواد المتفاعلة يسمى تفاعلاً من الدرجة الثانية Second order reaction

27
وبصفة عامة، فقد وجد من التجربة أن درجة التفاعل تتناسب مع الأس لتركيز المكونات المؤثرة في سرعة التفاعل ويمكن وضع هذا في الصيغة الرياضية التالية، ففي التفاعل: A + B → X سرعة التفاعل R تتناسب مع أس التراكيز كما يلي: R α CAm . CBn R = K CAm . CBn وهذا التفاعل تكون درجته m بالنسبة للمادة n , A بالنسبة للمادة B درجة التفاعل الكلية هي = n + m والصيغة العامة للتعبير عن سرعة التفاعل هي: R = -dC/dt = K CAm . CBn dCx/dt = K CAm . CBn

28
وهذا تفاعل من الدرجة الثانية.
وتعتمد سرعة التفاعل على معدل تكون المواد الناتجة أو استهلاك المواد المتفاعلة، أو بعبارة أخرى أدق على التركيز لكل مادة من المواد المشتركة في التفاعل بحيث يكون كل تركيز مرفوعاً إلى أس يمكن تعيينه بالتجربة. وهذا الأس قد يكون أعداداً صحيحة مثل 1، 2، 3،... وقد يكون كسراً من الواحد الصحيح، ومن الأمثلة على درجة التفاعل تدرس التفاعلات الآتية: 1.تفكك خامس أكسيد النيتروجين 2N2O5 → NO2 + O2 معادلة سرعة التفاعل dC(N2O5)/dt = K[N2O5] هذا تفاعل من الدرجة الأولى. 2 .تفكك ثاني أكسيد النيتروجين 2NO2 → NO +O2 معادلة سرعة التفاعل هي: dC(NO2)/dt = K[NO2]2 وهذا تفاعل من الدرجة الثانية.
/dt = K[N2O5]1 هذا تفاعل من الدرجة الأولى. 2 .تفكك ثاني أكسيد النيتروجين. 2NO2 → 2NO +O2. معادلة سرعة التفاعل هي: -dC(NO2)/dt = K[NO2]2. وهذا تفاعل من الدرجة الثانية.")
29
3.تفاعل ثلاثي إيثيل أمين مع بروميد الايثيل في محلول البنزين:
(C2H5)3N C2H5 Br (C2H5)4NBr بروميد رباعي إيثيل أمين ثلاثي إيثيل أمين معادلة سرعة التفاعل: -dC(C2H5Br)/dt = K[C2H5Br] [(C2H5)3N] هذا التفاعل كما يظهر من معادلة السرعة درجته الكلية هي الثانية ولكنه أيضاً تفاعل من الدرجة الأولى بالنسبة إلى مادة بروميد الايثيل وأيضاً من الدرجة الأولى بالنسبة إلى مادة ثلاثي إيثيل أمين. 4.تفكك الاستيالدهيد في الحالة الغازية، عند درجة 450ە م. CH3CHO CH4 + CO -dC(CH3CHO)/dt = K[CH3CHO]3/2 أي أن درجة التفاعل هي 3/2 .
3N + C2H5 Br (C2H5)4NBr. بروميد رباعي إيثيل أمين ثلاثي إيثيل أمين. معادلة سرعة التفاعل: -dC(C2H5Br)/dt = K[C2H5Br] [(C2H5)3N] هذا التفاعل كما يظهر من معادلة السرعة درجته الكلية هي الثانية ولكنه أيضاً تفاعل من الدرجة الأولى بالنسبة إلى مادة بروميد الايثيل وأيضاً من الدرجة الأولى بالنسبة إلى مادة ثلاثي إيثيل أمين. 4.تفكك الاستيالدهيد في الحالة الغازية، عند درجة 450ە م. CH3CHO CH4 + CO. -dC(CH3CHO)/dt = K[CH3CHO]3/2. أي أن درجة التفاعل هي 3/2 .")
30
وبذلك فإنه ليس من الضروري أن تساوي درجة التفاعل أعداداً صحيحة فقد تكون درجة التفاعل تساوي صفراً، أو نصفاً أو ثلاث أنصاف أو أعداداً صحيحة (1، 2، 3،....) وتحديدها يكون فقط من معادلة سرعة التفاعل واختيار ما يطابق المعادلة مع أس التركيز وعندما يكون التفاعل من الدرجة صفر فإن هذا يدل على أن التركيز لا يؤثر في سرعة التفاعل...كما أنه من المهم جداً تأكيد عدم وجود علاقة بين وزن المعادلة الكيميائية ودرجة التفاعل كما يتضح من المثالين الأول والثاني حيث أن لهما معادلات كيميائية متشابهة من ناحية الوزن الكيميائي بينما التفاعل في المثال الأول من الدرجة الأولى وفي المثال الثاني من الدرجة الثانية. يمكن ان يكون قانون سرعة التفاعل لا يعتمد على كل مادة متفاعلة او ناتجة

31
تفكك يود الهيدروجين إلى اليود والهيدروجين 2HI H2 + I2
الجزيئية ودرجة التفاعل: تتعلق الجزيئية بميكانيكية التفاعل من الوجهة النظرية، ولما كان التفاعل يمكن أن يتم في عدة خطوات منها السريع ومنها البطيء فإن سرعة التفاعل الكلية تحدد بسرعة أبطأ خطوة والتي تعرف بالخطوة المحددة لسرعة التفاعل ودرجته. وتعرف جزيئية التفاعل بعدد الذرات أو الجزيئات التي تشترك في التفاعل وهي دائماً عدد صحيح، بينما درجة التفاعل ليس ضرورياً أن تساوي أعداداً صحيحة...ولكن بالرغم من أنه ليس هناك ارتباط بين درجة التفاعل وجزئيته، فإنه من المحتمل أحياناً أن تكون جزيئية التفاعل مساوية لدرجته كما في المثال الآتي: تفكك يود الهيدروجين إلى اليود والهيدروجين 2HI H2 + I2 نجد هذا التفاعل من الدرجة الثانية لأن سرعته تتناسب مع (HI)2 الأس الثاني لتركيز يوديد الهيدروجين، كما أن هذا التفاعل ثنائي الجزيئية Bi-molecular لأن جزيئين من يوديد الهيدروجين يجب أن يصطدما ليتم التفاعل.
2 الأس الثاني لتركيز يوديد الهيدروجين، كما أن. هذا التفاعل ثنائي الجزيئية Bi-molecular لأن جزيئين من يوديد الهيدروجين يجب أن يصطدما ليتم التفاعل.")
32
ويعرف التفاعل أحادي الجزيئية بأنه ذلك التفاعل الذي ينقسم فيه جزئ واحد إلى أجزاء أصغر مثال:
Br Br أو قد يحدث في الجزئي تغيرات داخلية كما يحدث عندما يتحول حمض الماليك إلى حمض الفيوماريك أما التفاعل الثنائي فهو ذلك التفاعل الذي يلزم فيه اصطدام جزيئين من المادة قبل أن يحدث التفاعل، والتفاعل الثلاثي الجزيئية هو ذلك التفاعل الذي يجب فيه التقاء ثلاث جزيئات من المادة المتفاعلة في وقت واحد حتى يحدث التفاعل، إلا أن التفاعلات الكيميائية عادة لا تكون بالسهولة التي تبدو عليها في المعادلة الكيميائية لأن هناك عدداً من العوامل المعقدة تؤثر في التفاعل، كما أن المعادلة بصيغتها المكتوبة لا تعطي صورة كاملة لمجرى التفاعل، فنجد مثلاً أنه قد تتفاعل النواتج لتعطي المواد المتفاعلة ثانية كما في التفاعلات العكسية، أو يمكن أن يتفاعل جزء منها ليعطي مركبات أخرى كما في التفاعلات المتسلسلة، أو يحدث أحياناً أن يتم التفاعل عدة طرق كما في التفاعلات الجانبية

33
يلقي المثال التالي ضوءاً اكبر على إيضاح المقصود بالجزيئية:
يلقي المثال التالي ضوءاً اكبر على إيضاح المقصود بالجزيئية: برومة الأسيتون في محلول مائي مخفف كما في المعادلة الآتية: CH3COCH3 + Br CH3COCH2Br + HBr هذا التفاعل من الدرجة الأولى وثنائي الجزيئية والقياسات التجريبية أثبتت أن سرعة التفاعل تتناسب مع تركيز الأسيتون فقط ولا تعتمد على تركيز البروم (أي من الدرجة الأولى) ويشمل التفاعل الكلى عدة خطوات وأن الخطوة المحددة لسرعة التفاعل ودرجته لا يشترك فيها البروم والخطوات المقترحة للتفاعل كالتالي: CH3COCH CH3-C = CH2 OH CH3 – C = CH2 + Br CH3-CO-CH2Br + HBr Ketonic ذلك فإن سرعة التفاعل الكيميائي تحكمها الخطوة البطيئة لتغير الأسيتون من الشكل الكيـتوني Enolic إلى الشكل الاينولى .
 ويشمل التفاعل الكلى عدة خطوات وأن الخطوة المحددة لسرعة التفاعل ودرجته لا يشترك فيها البروم والخطوات المقترحة للتفاعل كالتالي: CH3COCH3 CH3-C = CH2. OH. CH3 – C = CH2 + Br2 CH3-CO-CH2Br + HBr. Ketonic ذلك فإن سرعة التفاعل الكيميائي تحكمها الخطوة البطيئة لتغير الأسيتون من الشكل الكيـتوني. Enolic إلى الشكل الاينولى .")
34
ثابت التفاعل النوعي: Specific Rate Reaction (K)
لدينا التفاعل التالي: نواتج A B سرعة التفاعل R تتناسب مع التركيز: R α CA . CB or R = K CA . CB حيث K ثابت التناسب ويسمى ثابت السرعة النوعي. ويمكن كتابة الصيغة العامة كما يأتي: R = KCn , K = R/Cn (5 ) وبصفة عامة فإن وحدات ثابت سرعة التفاعل K تعتمد على الوحدات المستعملة للتعبير عن سرعة التفاعل، أي أنه يعبر عنها بوحدة الوزن الجزئي مقسوماً على الثانية (وزن جزيئي/لتر.ثانية) أي: Moles / liter. Sec ويمكن كتابتها ML-1 Sec-1وعندما تكون درجة التفاعل هي (n) فإن وحدات ثابت سرعة التفاعل النوعي M1-n . Ln-1 Sec-1 وذلك بالتعويض في العلاقة K=R/Cn .
 وبصفة عامة فإن وحدات ثابت سرعة التفاعل K تعتمد على الوحدات المستعملة للتعبير عن سرعة التفاعل، أي أنه يعبر عنها بوحدة الوزن الجزئي مقسوماً على الثانية (وزن جزيئي/لتر.ثانية) أي: Moles / liter. Sec ويمكن كتابتها ML-1 Sec-1وعندما تكون درجة التفاعل هي (n) فإن وحدات ثابت سرعة التفاعل النوعي M1-n . Ln-1 Sec-1 وذلك بالتعويض في العلاقة K=R/Cn .")
35
وزن جزيئي/لتر/ثانية ML-1 Sec-1
وحدات ثابت سرعة التفاعل النوعي (K) درجة التفاعل وزن جزيئي/لتر/ثانية ML-1 Sec-1 n = 0 M1/2 L1/2 Sec-1 n = 1/2 Sec-1 n = 1 L1/2 M1/2 Sec-1 n = 3/2 L M-1 Sec-1 n = 2 L2 M-2 Sec-1 n= 3
 درجة التفاعل. وزن جزيئي/لتر/ثانية ML-1 Sec-1. n = 0. M1/2 L1/2 Sec-1. n = 1/2. Sec-1. n = 1. L1/2 M1/2 Sec-1. n = 3/2. L M-1 Sec-1. n = 2. L2 M-2 Sec-1. n= 3.")
36
قانون السرعة ا) أوجد قانون سرعة للتفاعل الأتى الذي يحدث عند درجة حرارة 25°Cمستعينا بالمعلومات بالجدول الاتي: ب) أحسب قيمة ثابت سرعة التفاعل جـ) حدد رتبة التفاعل الكلية 2NO(g) + O2(g) NO2(g)
 أوجد قانون سرعة للتفاعل الأتى الذي يحدث عند درجة حرارة 25°Cمستعينا بالمعلومات بالجدول الاتي: ب) أحسب قيمة ثابت سرعة التفاعل جـ) حدد رتبة التفاعل الكلية 2NO(g) + O2(g) 2NO2(g)")
37
التركيز الابتدائي للمتفاعلات
2NO(g) + O2(g) NO2(g) التركيز الابتدائي للمتفاعلات (mol/L) السرعة الابتدائية (mol/L*s) التجربة O2 NO 1 1.10x10-2 1.30x10-2 3.21x10-3 2 2.20x10-2 1.30x10-2 6.40x10-3 3 1.10x10-2 2.60x10-2 12.8x10-3 4 3.30x10-2 1.30x10-2 9.60x10-3 5 1.10x10-2 3.90x10-2 28.8x10-3
 + O2(g) 2NO2(g) التركيز الابتدائي للمتفاعلات. (mol/L) السرعة الابتدائية. (mol/L*s) التجربة. O2. NO x x x x x x x x x x x x x x x10-3.")
38
حركية التفاعلات البسيطة
هى التفاعلات التى لها الرتبة صفر ,واحد , اثنين وهكذا على اساس ان التفاعل يحدث عند : 1- درجة حرارة ثابتة 2- الحجم ثابت 3- التفاعل غير عكسي 4- جميع التفاعلات متجانسة 5- رتبة التفاعل تساوى الجزيئية

39
تفاعلات الرتبة الصفرية
هى التى سرعتها لا تتأثر بتغير تراكيز مادة أو أكثر من المواد المشتركة في التفاعل لأنها تعتمد على عوامل اخرى غير التركيز مثل كمية الضوء الممتص فى حالة التفاعلات الضوئية أو الحافز فى حالة التفاعلات الحفزية A → P فترة نصف العمر :هى الزمن اللازم لتفكك نصف تركيز المادة المتفاعلة

40
تفاعلات الرتبة الصفرية
rate = k [A]0 = k t=0تركيز المادة عند بداية الزمن [A]0 [A] - [A]0 = -kt tتركيز المادة عند اى زمن [A] t½ = t when [A] = [A]0/2 t½ = [A]0 2k

41
تفاعلات الرتبة الأولى [A] = (a – x) = a e-kt a 0 t = 0 a-x x t = t
وحدة ثابت سرعة التفاعل للرتبة الأولى sec-1 فترة نصف العمر لتفاعل احادى الرتبة t1/2 = ln2/k
![تفاعلات الرتبة الأولى [A] = (a – x) = a e-kt a 0 t = 0 a-x x t = t](http://slideplayer.ae/slide/15551337/88/images/41/%D8%AA%D9%81%D8%A7%D8%B9%D9%84%D8%A7%D8%AA+%D8%A7%D9%84%D8%B1%D8%AA%D8%A8%D8%A9+%D8%A7%D9%84%D8%A3%D9%88%D9%84%D9%89+%5BA%5D+%3D+%28a+%E2%80%93+x%29+%3D+a+e-kt+a+0+t+%3D+0+a-x+x+t+%3D+t.jpg "وحدة ثابت سرعة التفاعل للرتبة الأولى sec-1. فترة نصف العمر لتفاعل احادى الرتبة t1/2 = ln2/k.")
42
بالنسبة إلى المواد المتفاعلة :
أي أن سرعة التفاعل تتناسب مع تركيز المواد المتفاعلة: α CA R R = -dcA / dt = KC بترتيب المعادلة ينتج: -dC/C = K dt بتكامل المعادلة يتم الحصول على: - dC/ C = ∫ K dt حيث K- هي ثابت التكامل. لإيجاد ثابت التكامل، نجد أنه عند بداية التفاعل كان التركيز الأولي هو a أي عندما كان الزمن = صفر، بالتعويض في المعادلة رقم (9) ينتج أن :
 ينتج أن :")
43
- ln a = K- بالتعويض عن قيمة K- في المعادلة (9): - ln C = Kt –ln a -ln C + ln a= Kt ومنه ln a/C = Kt log a/C = Kt Or log a/C = Kt / ويمكن أن تكتب المعادلة بدلالة ثابت السرعة كما يلي: K= 1/t ln a/C K = 2.303/t log a/C وكثيراً ما تكتب المعادلة (11) بالصيغة الأسية الآتية : C = a e-Kt المعادلات (10)-(12) تعبر عن التفاعل من الدرجة الأولى بالنسبة إلى تركيز المواد المتفاعلة حيث تركيز المواد المتفاعلة يكون C .
: - ln C = Kt –ln a -ln C + ln a= Kt ومنه ln a/C = Kt log a/C = Kt Or log a/C = Kt / ويمكن أن تكتب المعادلة بدلالة ثابت السرعة كما يلي: K= 1/t ln a/C K = 2.303/t log a/C وكثيراً ما تكتب المعادلة (11) بالصيغة الأسية الآتية : C = a e-Kt المعادلات (10)-(12) تعبر عن التفاعل من الدرجة الأولى بالنسبة إلى تركيز المواد المتفاعلة حيث تركيز المواد المتفاعلة يكون C .")
44
2 .بالنسبة للمواد الناتجة:
سرعة التفاعل تتناسب مع تركيز المواد الناتجة فإذا كانت الكمية التي تكونت من المواد الناتجة بعد مضى زمن قدره t هي (x)، فإن تركيز المواد المتفاعلة (a) الأولى ينقص بالمقدار نفسه ويصبح تركيز الكمية الباقية (a - x). R = dx/dt = K(a-x) بترتيب المعادلة نجد أن: dx /(a-x) = Kdt بتكامل المعادلة يتم الحصول على : -ln(a-x) = Kt + K- حيث K΄ ثابت التكامل، ويمكن استنتاج قيمته بالتعويض عن التركيز في بداية التفاعل أي عندما تكون ( t = o) فإن (x= صفر) وتؤول المعادلة إلى : ln a = K΄
، فإن تركيز المواد المتفاعلة (a) الأولى ينقص بالمقدار نفسه ويصبح تركيز الكمية الباقية (a - x). R = dx/dt = K(a-x) بترتيب المعادلة نجد أن: dx /(a-x) = Kdt. بتكامل المعادلة يتم الحصول على : -ln(a-x) = Kt + K- حيث K΄ ثابت التكامل، ويمكن استنتاج قيمته بالتعويض عن التركيز في بداية التفاعل أي عندما تكون ( t = o) فإن (x= صفر) وتؤول المعادلة إلى : - ln a = K΄")
45
وبالتعويض عن قيمة K΄ يتم الحصول على : - ln (a-x) = Kt –ln a ln (a/a-x) = Kt 2.303log (a/a-x) = Kt log (a/a-x) = Kt / K = 1/t 1n (a/a-x) K = [ log (a/a-x)] /t ويمكن وضع المعادلة في الصيغة الأسية كالآتي : X = a (1 – e-Kt)
![وبالتعويض عن قيمة K΄ يتم الحصول على : - ln (a-x) = Kt –ln a ln (a/a-x) = Kt 2.303log (a/a-x) = Kt log (a/a-x) = Kt / K = 1/t 1n (a/a-x) K = [ log (a/a-x)] /t ويمكن وضع المعادلة في الصيغة الأسية كالآتي : X = a (1 – e-Kt)](http://slideplayer.ae/slide/15551337/88/images/45/%D9%88%D8%A8%D8%A7%D9%84%D8%AA%D8%B9%D9%88%D9%8A%D8%B6+%D8%B9%D9%86+%D9%82%D9%8A%D9%85%D8%A9+K%CE%84+%D9%8A%D8%AA%D9%85+%D8%A7%D9%84%D8%AD%D8%B5%D9%88%D9%84+%D8%B9%D9%84%D9%89+%3A+-+ln+%28a-x%29+%3D+Kt+%E2%80%93ln+a+ln+%28a%2Fa-x%29+%3D+Kt+2.303log+%28a%2Fa-x%29+%3D+Kt+log+%28a%2Fa-x%29+%3D+Kt+%2F+K+%3D+1%2Ft+1n+%28a%2Fa-x%29+K+%3D+%5B+log+%28a%2Fa-x%29%5D+%2Ft+%D9%88%D9%8A%D9%85%D9%83%D9%86+%D9%88%D8%B6%D8%B9+%D8%A7%D9%84%D9%85%D8%B9%D8%A7%D8%AF%D9%84%D8%A9+%D9%81%D9%8A+%D8%A7%D9%84%D8%B5%D9%8A%D8%BA%D8%A9+%D8%A7%D9%84%D8%A3%D8%B3%D9%8A%D8%A9+%D9%83%D8%A7%D9%84%D8%A2%D8%AA%D9%8A+%3A+X+%3D+a+%281+%E2%80%93+e-Kt%29.jpg "وبالتعويض عن قيمة K΄ يتم الحصول على : - ln (a-x) = Kt –ln a ln (a/a-x) = Kt 2.303log (a/a-x) = Kt log (a/a-x) = Kt / K = 1/t 1n (a/a-x) K = [ log (a/a-x)] /t ويمكن وضع المعادلة في الصيغة الأسية كالآتي : X = a (1 – e-Kt)")
46
تفاعلات الرتبة الأولى ln[A] = ln[A]0 - kt
![تفاعلات الرتبة الأولى ln[A] = ln[A]0 - kt](http://slideplayer.ae/slide/15551337/88/images/46/%D8%AA%D9%81%D8%A7%D8%B9%D9%84%D8%A7%D8%AA+%D8%A7%D9%84%D8%B1%D8%AA%D8%A8%D8%A9+%D8%A7%D9%84%D8%A3%D9%88%D9%84%D9%89+ln%5BA%5D+%3D+ln%5BA%5D0+-+kt.jpg "تفاعلات الرتبة الأولى ln[A] = ln[A]0 - kt")
47
ا) ثابت سرعة التفاعل ب) فترة نصف العمر
مثال: إذا كان مركب يتفكك حسب تفاعل من الرتبة الأولى ووجد أن 20% منه تفكك فى زمن قدره 22 دقيقة فما هو: ا) ثابت سرعة التفاعل ب) فترة نصف العمر ج) ما النسبة المتبقية من المادة المتفاعلة بعد خمس ساعات a = 100 , x =100*0.2 =20 k =1/22 ln 100/80 = 0.01 min-1 t ½ = /k = min ln (a-x)/a = -kt = -5*60*0.01= -3 (a-x)/a = exp(-3) = 0.05 = 5%
 ثابت سرعة التفاعل ب) فترة نصف العمر. ج) ما النسبة المتبقية من المادة المتفاعلة بعد خمس ساعات. a = 100 , x =100*0.2 =20. k =1/22 ln 100/80. = 0.01 min-1. t ½ = 0.693/k. = 69.3 min. ln (a-x)/a = -kt = -5*60*0.01= -3. (a-x)/a = exp(-3) = 0.05 = 5%")
48
تفاعلات الرتبة الأولى الظاهرية
هى تفاعلات تحقق معادلة الرتبة الأولى مع اعتبار ان اكثر من مادة متفاعلة تكون في التفاعل وان وجود تركيز مادة أو أكثر من مواد التفاعل بكمية زائدة في حيز التفاعل يبقى تركيزها ثابتا تقريبا خلال التفاعل مما يؤدى إلى أن سرعة التفاعل لا تتأثر بشكل ملحوظ بهذا التركيز أو التراكيز مثال أى ان سرعة التفاعل تتأثر فقط بتركيز الخلات لذا فإن رتبة التفاعل أولى والنتائج العملية تؤكد هذا.

49
أ- أمثلة على تفاعلات الرتبة الأولى في المحلول:
تحويل -N كلورواستتانيلايد إلى بارا – كلورو استتانيلايد: يعد هذا التفاعل من الدرجة الأولى ويمكن كتابة معادلة التفاعل كالتالي: ويتابع سير التفاعل بأخذ حجم متساوي من التفاعل في فترات متفاوتة، ثم يضاف في كل مرة زيادة من يوديد البوتاسيوم ويعاير اليود المتصاعد بمحلول قياسي من الثيوكبربتات، ويلاحظ أن المركب (1) يستطيع أن يطرد اليود من يوديد البوتاسيوم بينما المركب (2) لا يستطيع ذلك، وبهذا تكون المعايرة قياساً لتركيز الكميات الباقية من المواد المتفاعلة، بينما تكون معايرة الحجم نفسه من المحلول قبل تحويله قياسياً للتركيز الابتدائي، ولذلك يمكن حساب ثابت سرعة التفاعل النوعي في كل مرة باستخدام المعادلة : K = [2.303/t] log a/(a-x) أو المعادلة log (a/a- x) = [Kt ]/
 يستطيع أن يطرد اليود من يوديد البوتاسيوم بينما المركب (2) لا يستطيع ذلك، وبهذا تكون المعايرة قياساً لتركيز الكميات الباقية من المواد المتفاعلة، بينما تكون معايرة الحجم نفسه من المحلول قبل تحويله قياسياً للتركيز الابتدائي، ولذلك يمكن حساب ثابت سرعة التفاعل النوعي في كل مرة باستخدام المعادلة : K = [2.303/t] log a/(a-x) أو المعادلة log (a/a- x) = [Kt ]/")
50
- التفكك المحفز لفوق أكسيد الهيدروجين : H2O2 → H2O + 1/2 O2 يمكن تتبع بسحب عينات من خليط التفاعل عند أزمنه مختلفة ثم معايرتها بمحلول معلوم العيارية من برمنـجنات البوتاسيوم. الحجم (V) مل المأخوذ من محلول البرمنجنات يكون مكافئاً لكمية فوق أكسيد الهيدروجين غير المفككة أي مكافئاً للمقدار ( a- x) وبرسم log v مقابل الزمــــن t يتم الحصول على خــط مسـتقيم ومن ميله يمكن تعيين قيمة ثابت سرعة التفاعل - تفاعلات الانحلال النووي: كل تفاعلات الانحلال النووي أحادية الرتبة ،وتسري عليها جميع معادلات وقوانين الرتبة الأولى بما فيها معادلة زمن نصف العمر، وزمن نصف العمر للمواد المشعة ذات أهمية قصوى في الكيمياء الإشعاعية
 مل المأخوذ من محلول البرمنجنات يكون مكافئاً لكمية فوق أكسيد الهيدروجين غير المفككة أي مكافئاً للمقدار ( a- x) وبرسم log v مقابل الزمــــن t يتم الحصول على خــط مسـتقيم ومن ميله يمكن تعيين قيمة ثابت سرعة التفاعل - تفاعلات الانحلال النووي: كل تفاعلات الانحلال النووي أحادية الرتبة ،وتسري عليها جميع معادلات وقوانين الرتبة الأولى بما فيها معادلة زمن نصف العمر، وزمن نصف العمر للمواد المشعة ذات أهمية قصوى في الكيمياء الإشعاعية .")
51
تفاعلات الرتبة الأولى الكاذبة:
وهذه تكون تفاعلات ثنائية الجزيئية، حيث فيها أحد المكونات يكون متواجدا بكميات كبيرة مثل لو كان المذيب أحد المتفاعلات فبالرغم من كون التفاعل حينئذ ثنائي الجزيئية، إلا أن النتائج التجريبية ستخضع لقانون الرتبة الأولى، حيث يكون من المستحيل أن يلاحظ التغير الطفيف الحادث في تركيز المكون الزائد نتيجة للتفاعل. فمثلاً مولارية الماء = = مول 18جم/مول أي أنه لا يمكن ملاحظة النقص في تركيز الماء إذا بدء التفاعل بتركيز من المواد المتفاعلة مولار =

52
1 - تميؤ خلات الإيثيل: إذا تحلل مائياً وبصورة كاملة 0
1 - تميؤ خلات الإيثيل: إذا تحلل مائياً وبصورة كاملة 0.1 مول من محلول خلات الايثيل تبعاً للمعادلة : CH3COOC2H5 + H2O → CH3 COOH + C2H5OH فإن النقص في مولارية الماء سيكون 0.1 مول لكل مول، بمعنى أن النقص سيكون مساويا 0.18 أي سيكون أقل من 0.2% هذا يعني أن سرعة التفاعل لا تعتمد على التركيز الابتدائي للماء ولكنها تعتمد فقط على تركيز خلات الايثيل، أي أنه يتبع قانون الرتبة الأولى للتفاعلات.

53
2 - التميؤ الحامضي لخلات المثيل: CH3COOCH3 + H2O → CH3 COOH + CH3OH هذا التميؤ يحدث في وجود زيادة من الماء، وفي وجود حمض مخفف كعامل حفز، وهو تفاعل أحادي الرتبة ، ذلك لأن سرعة التفاعل تعتمد فقط على تركيز الإستر، ولا تعتمد على تركيز أيون الهيدروجين حيث أن الماء موجود بكمية كبيرة يمكن معها افتراض أن تركيزها ثابت أثناء سير التفاعل. ويمكن تتبع هذا التفاعل بأخذ عينات من الخليط من وقت لآخر ومعايرتها بمحلول قلوي قياسي. ويكون حجم القلوي المستخدم في كل معايرة مكافئاً لكمية حامض الخليك الناتجة من التميؤ أي مكافئة لـ (x) ويكون الحجم المأخوذ من القلوي بعد تمام التميؤ ممثلاً لكمية التركيز الابتدائي (a) وتطبق العلاقة: K=2.303/t log a/(a-x) أو العلاقة log (a/a-x) = Kt / 2.303
 ويكون الحجم المأخوذ من القلوي بعد تمام التميؤ ممثلاً لكمية التركيز الابتدائي (a) وتطبق العلاقة: K=2.303/t log a/(a-x) أو العلاقة log (a/a-x) = Kt /")
54
أيضاً التحليل المائي لمركب كلوريد البيوتيل الثـلثي في الوسط الحمضي يعتبر من الأمثلة النموذجية على تفاعلات أحادية الرتبة الكاذبة لان سرعة التفاعل تعتمد فقط على تركيز كـلوريد الألكيل ولا تتأثر بتركيز الماء. ولذلك ليس ضرورياً أن يكون تركيز الماء بكميات زائدة حيث يجري التفاعل في وجود حمض الفورميك والماء. 3- تحول سكر القصب إلي سكر عنب: معادلة التفاعل هي : C12H22On + H2O → 2C6H12O6 سكر عنب (جلوكوز) سكر القصب (سكروز) في هذا التفاعل كل من سكر القصب والماء يدخل في التفاعل، ولكن سرعة التفاعل تعتمد على تركيز سكر القصب ولذلك فالتفاعل من الدرجة الأولى. ويمكن حساب ثابت التفاعل النوعي من المعادلتين السابقتين: K= (2.303/t) log a/(a-x) log (a/a-x) = Kt / (2.303) وسبب أن هذا التفاعل من الدرجة الأولى رغم وجود أكثر من مادة تدخل في التفاعل هو الماء موجود بكميات زائدة أن تركيز لا يتغير تغيراً كبيراً خلال التفاعل والتغير الطفيف الذي يحدث على الماء جوداً بكميات قليلة بحيث يتغير تركيزه بمرور التفاعل فإن درجة التفاعل تكون من الدرجة الثانية.
 سكر القصب (سكروز) في هذا التفاعل كل من سكر القصب والماء يدخل في التفاعل، ولكن سرعة التفاعل تعتمد على تركيز سكر القصب ولذلك فالتفاعل من الدرجة الأولى. ويمكن حساب ثابت التفاعل النوعي من المعادلتين السابقتين: K= (2.303/t) log a/(a-x) log (a/a-x) = Kt / (2.303) وسبب أن هذا التفاعل من الدرجة الأولى رغم وجود أكثر من مادة تدخل في التفاعل هو الماء موجود بكميات زائدة أن تركيز لا يتغير تغيراً كبيراً خلال التفاعل والتغير الطفيف الذي يحدث على الماء جوداً بكميات قليلة بحيث يتغير تركيزه بمرور التفاعل فإن درجة التفاعل تكون من الدرجة الثانية.")
55
ثانياً : التفاعل من الدرجة الثانية : عندما تتناسب سرعة التفاعل مع الأس الأول لتركيز كل من المادتين المتفاعلتين فإن التفاعل يكون الدرجة الثانية، فإذا افترض أن هناك تفاعلاً وكان التركيز الأولى لإحدى المواد هو (a) وللأخرى هي (b) فلإيجاد معادلة سرعة التفاعل هناك احتمالان هما : 1- أن تكون المادتان المتفاعلتان لهما نفس التركيز أي أن : a = b وسرعة التفاعل تتناسب مع ناتج هذا التفاعل. 2- أن تكون المادتان المتفاعلتان لهما تركيزين مختلفين أي أن : a ≠ b وسرعة التفاعل تتناسب مع ناتج هذا التفاعل.

56
في حالة الاحتمال الأول، أي عندما تكون المادتان المتفاعلتان ذواتي تركيز متساو
A B rate = k [A]2 tتركيز المادة عند اى زمن (a-x) t=0تركيز المادة عند بداية الزمن (a) فترة نصف العمر لتفاعل ثنائى الرتبة t½ = t when x = a /2 t½ = 1 a
 t=0تركيز المادة عند بداية الزمن (a) فترة نصف العمر لتفاعل ثنائى الرتبة. t½ = t when x = a /2. t½ = 1. a.")
57
تفاعلات الرتبة الثانية

58
إيجاد معادلة سرعة التفاعل من الدرجة الثانية في حالة كون المادتان المتفاعلتان لهما تركيزين مختلفتين
A + B → نواتج فإذا كان تركيز المادة A هو a، وتركيز المادة B هو b، وحيث أن : a ≠ b فإذا كان الجزء الناتج من التفاعل عند t هو x. :. الجزء المتبقي من المادة A هو : (a - x) والجزء المتبقي من المادة B هو : (b - x) وبالتعويض في معادلة السرعة للتفاعل من الدرجة الثانية : dx/dt = K (a-x) (b-x) وبترتيب المعادلة نحصل على: dx/(a-x) (b-x) = K dt باستخدام التكامل التجزيء:
 والجزء المتبقي من المادة B هو : (b - x) وبالتعويض في معادلة السرعة للتفاعل من الدرجة الثانية : dx/dt = K (a-x) (b-x) وبترتيب المعادلة نحصل على: dx/(a-x) (b-x) = K dt. باستخدام التكامل التجزيء:")
59
باعتبار (b-x) = مقدار ثابت = B تؤول المعادلة إلى :
dx/dt = KB(a-x) dx/(a-x) = KB dt بالتكامل: ∫dx/(a-x) = ∫KB dt -ln(a-x) = KBt + K- t = o فإن x = o عندما K = ln a ln a/(a-x) = KBt باعتبار (a -x ) مقدار ثابت = A تؤول المعادلة إلى الشكل : dx/dt = KA (b-x) dx/(b-x) = KAdt ln b/(b-x) = Kat بطرح المعادلة (23) من المعادلة (24) والتعويض على A وB بما يساويهما ينتج أن : ln b/(b-x) – ln a/(a-x) = Kat – KBt ln b(a-x)/ a(b-x) = (a-b) Kt LM-1 Sec-1 (25)
 dx/(a-x) = KB dt. بالتكامل: ∫dx/(a-x) = ∫KB dt. -ln(a-x) = KBt + K- t = o فإن x = o عندما. K = ln a. ln a/(a-x) = KBt. باعتبار (a -x ) مقدار ثابت = A تؤول المعادلة إلى الشكل : dx/dt = KA (b-x) dx/(b-x) = KAdt. ln b/(b-x) = Kat. بطرح المعادلة (23) من المعادلة (24) والتعويض على A وB بما يساويهما ينتج أن : ln b/(b-x) – ln a/(a-x) = Kat – KBt. ln b(a-x)/ a(b-x) = (a-b) Kt LM-1 Sec-1 (25)")
60
أمثلة على التفاعل من الدرجة الثانية :
(1) تصبن خلات الإيثيل يمكن إجراء هذا التفاعل في وجود أيونات الهيدروكسيل (OH) على أن تكون بكميات قليلة حتى يتأثر تركيزها، وبذلك تكون درجة التفاعل هي الدرجة الثانية، أما وجود كميات كبيرة من الهيدروكسيل بحيث لا يتأثر تركيزها فإن التفاعل يكون من الدرجة الأولى، ويمكن كتابة معادلة التفاعل الصيغة الآتية : CH3COOC2H5+OH- → CH3COO- + C2H5OH وتكون متابعة سير التفاعل بأخذ كميات معلومة من خليط التفاعل في فترات متتالية ثم معايرتها بحمض قياسي، ويمكن حساب ثابت التفاعل من رسم العلاقة بين : t # x/(a-x) حيث Ka هي ميل الخط المستقيم الناتج.
 تصبن خلات الإيثيل. يمكن إجراء هذا التفاعل في وجود أيونات الهيدروكسيل (OH) على أن تكون بكميات قليلة حتى يتأثر تركيزها، وبذلك تكون درجة التفاعل هي الدرجة الثانية، أما وجود كميات كبيرة من الهيدروكسيل بحيث لا يتأثر تركيزها فإن التفاعل يكون من الدرجة الأولى، ويمكن كتابة معادلة التفاعل الصيغة الآتية : CH3COOC2H5+OH- → CH3COO- + C2H5OH. وتكون متابعة سير التفاعل بأخذ كميات معلومة من خليط التفاعل في فترات متتالية ثم معايرتها بحمض قياسي، ويمكن حساب ثابت التفاعل من رسم العلاقة بين : t # x/(a-x) حيث Ka هي ميل الخط المستقيم الناتج.")
61
(2) أكسدة حمض يوديد الهيدروجين بواسطة فوق أكسيد الهيدروجين في الوسط الحمضي :
يمكن كتابة معادلة التفاعل: 2Hl + H2O2 → l2 + 2H2O هذا التفاعل من الدرجة الثانية ويتم في عدة خطوات كالتالي : l- + H2O → H2O + lO- H + + lO- → HIO HlO + H+ + l- → H2O + l2 والخطوة الأولى هي الخطوة البطيئة وهي التي تحدد سرعة التفاعل ودرجته وعليه يمكن كتابة معادلة سرعة التفاعل كما يلي : [H2O2 ] [HI] = Rate of reaction = K R = سرعة التفاعل

62
التفاعلات الثنائية الكاذبة:
التفاعلات التي ليست من الدرجة الثانية حقيقة، ولكن تكون كذلك تحت شروط خاصة وتتغير درجتها بتغير هذه الشروط تسمى تفاعلات ثنائية كاذبة، فمثلا تأثير الكحولات على الأحماض العضوية بكميات زائدة من الكحول ، كما في المعادلة التالية: R – COOH + R΄ OH → R - COOR΄ + H2O في هذا التفاعل نجد أن سرعة التفاعل تعتمد على التركيز الحامض العضوي (RCOOH) كما أن هذا الحامض يقوم بدور الحافز لهذا التفاعل، وتركيزه يتغير في أثناء التفاعل ولذلك يعبر عن سرعة التفاعل كالتالي: سرعة التفاعل = ثابت x 2[R - COOH] فالتفاعل من الدرجة الثانية بالرغم من مشاركة أكثر من جزيئين في التفاعل في كل لحظة. وعندما بضاف حامض معدني ليقوم بدور الحافز ويظل تركيزه ثابتاً لا يتغير أثناء التفاعل فإن درجة هذا التفاعل تصبح من الدرجة الأولى.
 كما أن هذا الحامض يقوم بدور الحافز لهذا التفاعل، وتركيزه يتغير في أثناء التفاعل ولذلك يعبر عن سرعة التفاعل كالتالي: سرعة التفاعل = ثابت x 2[R - COOH] فالتفاعل من الدرجة الثانية بالرغم من مشاركة أكثر من جزيئين في التفاعل في كل لحظة. وعندما بضاف حامض معدني ليقوم بدور الحافز ويظل تركيزه ثابتاً لا يتغير أثناء التفاعل فإن درجة هذا التفاعل تصبح من الدرجة الأولى.")
63
ثالثا: التفاعل من الدرجة الثالثة:
التفاعلات الكيميائية التي درجتها أعلى من الدرجة الثانية أحياناً تكون مهمة ولو أنها نادرة، ويمكن وضع المعادلة الآتية للتفاعل ثلاث الدرجة : مواد ناتجة A + B + D → إيجاد معادلة سرعة التفاعل من الدرجة الثالثة بالنسبة إلى المواد الناتجة عندما تكون المواد المتفاعلة متساوية في التركيز. فإذا كان التركيز الابتدائي للمواد المتفاعلة هو a، فإنه بعد مضى زمن قدره t يكون تركيز المواد الناتجة هو x. .: معادلة سرعة التفاعل هي : R = K CA . AB . CD ولكن CA = CB = CD = a dx/dt = K (a-x) (26) بترتيب المعادلة نحصل على : ∫dx/(a-x)3 = ∫ K dt 1/2(a-x)2 = Kt + K- حيث K΄ هي ثابت التكامل، وتوجد قيمتها عندما تكون t = o، أي عند بداية التفاعل x = o :. K = 1/ 2a2 وبالتعويض عن قيمة بقيمتها نحصل على 1/2(a-x)2 = Kt + 1/2a2 Kt = 1/2(a-x)2 – 1/2a2 K = 1/2t (1/(a-x)2 – 1/a2) L2M-2Sec (27)
3 (26) بترتيب المعادلة نحصل على : ∫dx/(a-x)3 = ∫ K dt. 1/2(a-x)2 = Kt + K- حيث K΄ هي ثابت التكامل، وتوجد قيمتها عندما تكون t = o، أي عند بداية التفاعل x = o. :. K = 1/ 2a2. وبالتعويض عن قيمة بقيمتها نحصل على. 1/2(a-x)2 = Kt + 1/2a2. Kt = 1/2(a-x)2 – 1/2a2. K = 1/2t (1/(a-x)2 – 1/a2) L2M-2Sec-1 (27)")
64
من أهم التفاعلات الغازية من الدرجة الثالثة تفاعلات أول أكسيد النيتروجين مع الأكسجين أو البرومين أو الكلورين ويمكن وضع معادلات التفاعل كالتالي: 1) 2NO + O2 → 2NO2 2) 2NO + Br2 → 2NOBr 3) 2NO + Cl2 → 2NOCl ومعادلة سرعة التفاعل للتفاعلات السابقة هي : -d(NO) / dt = K(NO)2(X2) حيث تمثل تركيز الأكسجين أو البرومين أو الكلورين . وتعطي قيمة آ للتفاعلات الدرجة الثالثة عند a = b = c كما يلي: K=1/2τ [1/(a-a/2)2 – 1/a2] = 1/2τ.3/a2 τ =3/2 1/Ka2 Sec.
 2NO + O2 → 2NO2. 2) 2NO + Br2 → 2NOBr. 3) 2NO + Cl2 → 2NOCl. ومعادلة سرعة التفاعل للتفاعلات السابقة هي : -d(NO) / dt = K(NO)2(X2) حيث تمثل تركيز الأكسجين أو البرومين أو الكلورين . وتعطي قيمة آ للتفاعلات الدرجة الثالثة عند a = b = c كما يلي: K=1/2τ [1/(a-a/2)2 – 1/a2] = 1/2τ.3/a2. τ =3/2 1/Ka2 Sec.")
65
خامسا: الحالة العامة: لإيجاد درجة التفاعل في الحالة العامة أي عندما تكون درجة التفاعل (n) مع الأخذ في الاعتبار أن المواد المتفاعلة متساوية في تركيزها الأولى نتبع الطريقة التالية: R α Cn R = K (a – x )n dx/dt = K(a-x)n بفصل المتغيرات : dx/(a-x)n= K dt بإجراء عملية التكامل مع الأخذ في الاعتبار أن n ≠ 1 يتم الحصول على : ∫ dx/ (a- x)n = ∫K dt 1/ (n -1)(a-x)(n-1) = Kt + K΄ (29) حيث K΄ ثابت التكامل ويمكن استنتاج قيمته كما يلي : at t = o x = o K΄ = 1/ (n-1)(a-o)(n-1) = 1/ (n-1) a(n-1) وبالتعويض عن قيمة في المعادلة (29) نحصل على : 1/ (n-1)(a-x)(n-1) = Kt + (n-1) a(n-1) K= 1/ t(n-1) [1/ (a-x)(n-1) - 1/a(n-1)] K΄
 مع الأخذ في الاعتبار أن المواد المتفاعلة متساوية في تركيزها الأولى نتبع الطريقة التالية: R α Cn. R = K (a – x )n. dx/dt = K(a-x)n. بفصل المتغيرات : dx/(a-x)n= K dt. بإجراء عملية التكامل مع الأخذ في الاعتبار أن n ≠ 1 يتم الحصول على : ∫ dx/ (a- x)n = ∫K dt. 1/ (n -1)(a-x)(n-1) = Kt + K΄ (29) حيث K΄ ثابت التكامل ويمكن استنتاج قيمته كما يلي : at t = o x = o. K΄ = 1/ (n-1)(a-o)(n-1) = 1/ (n-1) a(n-1) وبالتعويض عن قيمة في المعادلة (29) نحصل على : 1/ (n-1)(a-x)(n-1) = Kt + (n-1) a(n-1) K= 1/ t(n-1) [1/ (a-x)(n-1) - 1/a(n-1)] K΄")
66
وبتطبيق معادلة الحالة العامة للتفاعل من الدرجة الثانية أي أن (n=2) نجد أن المعادلة تؤول إلى:
K = [1/t ] x/a(a-x) LM-1 Sec-1 وفي حالة التفاعل من الدرجة الثالثة أي أن (n=3) نجد أنها تؤول إلى : K = 1/2t [1/(a-x)2 – 1/a2] L2 M-2 Sec-1 وفي حالة التفاعل من الدرجة ثلاث أنصاف أي أن (n= 3/2) نجد أنها تؤول إلى : K = 2/t [1/ (a -x)1/2 - 1/a1/2] L1/2 M-1/2 Sec-1 وفي حالة التفاعل من الدرجة ثلاث أنصاف أي أن (n= ½) نجد أنها تؤول إلى : K = 2/t [ a½ - (a –x) ½]M½. L-½ .Sec-1
 LM-1 Sec-1. وفي حالة التفاعل من الدرجة الثالثة أي أن (n=3) نجد أنها تؤول إلى : K = 1/2t [1/(a-x)2 – 1/a2] L2 M-2 Sec-1. وفي حالة التفاعل من الدرجة ثلاث أنصاف أي أن (n= 3/2) نجد أنها تؤول إلى : K = 2/t [1/ (a -x)1/2 - 1/a1/2] L1/2 M-1/2 Sec-1. وفي حالة التفاعل من الدرجة ثلاث أنصاف أي أن (n= ½) نجد أنها تؤول إلى : K = 2/t [ a½ - (a –x) ½]M½. L-½ .Sec-1.")
67
تحليل النتائج الحركية تجرى التفاعلات الكيميائية بشكل عام عند درجة حرارة ثابتة حيث يتم تحضير مزيج معروف التركيب من المواد المتفاعلة فى وعاء للتفاعل وتثبت درجة الحرارة ثم يقاس الإنخفاض فى تركيز إحدى المواد المتفاعلة مع الزمن بالطرق المناسبةونرسم العلاقة بين التركيز والزمن او الزيادة فى تركيز احدى المواد الناتجة. لمتابعة تغير التركيز مع الزمن يكون ذلك بإحدى الطرق 1- التحليل الكيميائي 2- التحليل الفيزيائي التحليل الكيميائي : يمكن تقدير مباشرة بالطرق الاتية : الطريقة الحجمية ب) الطريقة الوزنية الطرق الفيزيائية : ا) قياس الضغط فى التفاعلات الغازية ب) قياس التمدد أو التغير فى الحجم ج) الطرق الضوئية (الإستقطاب-الإنكسار –الألوان –الطيف) د) الكهربية (التوصيل –فرق الجهد-المطياف الكتلى )
 الطريقة الوزنية. الطرق الفيزيائية : ا) قياس الضغط فى التفاعلات الغازية. ب) قياس التمدد أو التغير فى الحجم. ج) الطرق الضوئية (الإستقطاب-الإنكسار –الألوان –الطيف) د) الكهربية (التوصيل –فرق الجهد-المطياف الكتلى )")
68
الطريقة التفاضلية لإيجاد رتبة التفاعل
تعتمد هذه الطريقة على التعامل مع سرعة التفاعل مباشرة حيث يتم تقديرها عن طريق قياس ميول مماسات على منحــــــنيات التركيز –الزمن

69
تعيين رتبة وثابت التفاعل
وتطبق الطريقة التفاضلية على وجهين مختلفين 1- طريقة السرعة الإبتدائية.(Initial rate method) باجراء التفاعل عدة مرات باستخدام تراكيز ابتدائية مختلفة وتعيين السرعات الإبتدائية بقياس تغير التركيز مع الزمن فى كل تجربة وتطبق المعادلة السرعة الإبتدائية [A]t t / s
 باجراء التفاعل عدة مرات باستخدام تراكيز ابتدائية مختلفة وتعيين السرعات الإبتدائية بقياس تغير التركيز مع الزمن فى كل تجربة وتطبق المعادلة. السرعة الإبتدائية. [A]t. t / s.")
70
تعيين رتبة وثابت التفاعل
2- طريقة السرعة اللحظية: Instantaneous method تجرى تجربة واحدة يسجل فيها منحنى التركيز مع الزمن ثم نرسم مماسات متعددة عند نقاط مختلفة على المنحنى ( التركيز –الزمن ) حيث يمثل ميل المماس عند نقطة معينة السرعة اللحظية عند ذلك التركيزثم برسم علاقة بين لوغاريتم معدل سرعة التفاعل مع لوغاريتم التركيز ويكون ميل الخط مساويا α.ويلاحظ فى هذه الطريقة ان جميع المواد الأخرى يجب ان تكون فى زيادة حتى نلغى تأثيرها. ثم تعاد التجربة لإيجاد الرتبة β بشرط ان يكون تركيز كل من المواد الأخرى ثابتا
 حيث يمثل ميل المماس عند نقطة معينة السرعة اللحظية عند ذلك التركيزثم برسم علاقة بين لوغاريتم معدل سرعة التفاعل مع لوغاريتم التركيز ويكون ميل الخط مساويا α.ويلاحظ فى هذه الطريقة ان جميع المواد الأخرى يجب ان تكون فى زيادة حتى نلغى تأثيرها. ثم تعاد التجربة لإيجاد الرتبة β بشرط ان يكون تركيز كل من المواد الأخرى ثابتا.")
71
السرعة اللحظية = السرعة عند لحظة معينة
Br2 (aq) + HCOOH (aq) Br- (aq) + 2H+ (aq) + CO2 (g) ميل المماس ميل المماس ميل المماس متوسط السرعة= - D[Br2] Dt = - [Br2]2 – [Br2]1 t2 – t1 السرعة اللحظية = السرعة عند لحظة معينة
 + HCOOH (aq) 2Br- (aq) + 2H+ (aq) + CO2 (g) ميل المماس. ميل المماس. ميل المماس. متوسط السرعة= - D[Br2] Dt. = - [Br2]2 – [Br2]1. t2 – t1. السرعة اللحظية = السرعة عند لحظة معينة.")
72
تعيين رتبة وثابت التفاعل
3- طريقة التكامل Integration method تعتمد هذه الطريقة على رسم المعادلات التكاملية للرتب المختلفة حيث أن الرتبة الصحيحة للتـــفاعل ستعطي خـــطا مستقيما وأما الأخرى ستعطي منحنيات

73
الطريقة التكاملية لإيجاد رتبة التفاعل
1/[A]t = kt + 1/[A]0 [A]t = -kt + [A]0 ln[A]t = -kt + ln[A]0

74
تعيين رتبة وثابت التفاعل
4- طريقة عمر النصف Half –life period)) تعتمد هذه الطريقة على قياس الزمن اللازم لإختفاء نصف تركيز المواد المتفاعلة [A]0 concentration t1/2 [A]0/2 [A]0/4 t1/2 t1/2 [A]0/8 time
) تعتمد هذه الطريقة على قياس الزمن اللازم لإختفاء نصف تركيز المواد المتفاعلة. [A]0. concentration. t1/2. [A]0/2. [A]0/4. t1/2. t1/2. [A]0/8. time.")
77
6- طريقة فانت هوف التفاضلية :
في التفاعلات الآتية التي سرعتها R وتتضمن مادة واحدة متفاعلة تتناسب درجتها مع الأس n للتركيز تكون معادلة التفاعل لها كالتالي: R = dc/dt = KCn فإذا أجريت تجربتان وكان التركيز الابتدائي في التجربة الأولي هي c1 عند t1 وفي التجربة الثانية هي c2 عند t2 فإنه باستخدام طريقة التفاضل نحصل على : R1 =-dc1/dt1 = KC1n R2 = -dc2/dt2 = KC2n بقسمة المعادلتين ينتج أن: R1/R2 = K C1n/K C2n = (C1/C2)n logR1 – logR2 = n(logC1-logC2) n = logR1- logR2/ logC1-logC بما يساويها R وبالتعويض عن قيمة n= logC2/t2 – logC1/t1 / logC1-logC2
n. logR1 – logR2 = n(logC1-logC2) n = logR1- logR2/ logC1-logC2 بما يساويها R وبالتعويض عن قيمة. n= logC2/t2 – logC1/t1 / logC1-logC2.")
78
تعيين رتبة وثابت التفاعل
7- طريقة العزل Isolation Method)) تستخدم إذا وجد اكثر من مادة تؤثر على سرعة التفاعل .وهى تعتمد على جعل جميع تراكيز المواد المتفاعلة فيما ماعدا واحدة توجد بكميات زائدة فى حيز التفاعل ويؤدى هذا الى عدم تغيرها بشكل تركيز المادة الموجودة بكمية قليلة وبالتالى فان سرعة التفاعل سوف تتأثر فقط بتركيز تلك المادة الموجودة بكمية قليلة فى حيز التفاعل .
) تستخدم إذا وجد اكثر من مادة تؤثر على سرعة التفاعل .وهى تعتمد على جعل جميع تراكيز المواد المتفاعلة فيما ماعدا واحدة توجد بكميات زائدة فى حيز التفاعل ويؤدى هذا الى عدم تغيرها بشكل تركيز المادة الموجودة بكمية قليلة وبالتالى فان سرعة التفاعل سوف تتأثر فقط بتركيز تلك المادة الموجودة بكمية قليلة فى حيز التفاعل .")
79
تطبيق على القياسات الحركية استخدام القياسات الحركية في اقتراح ميكانيكيات للتفاعلات الكيميائية
اقتراح "ميكانيكية لتفاعل الهكسانون الحلقي مع صبغة الملاكيت الخضراء" (تفاعل مظلم محفز ضوئياً) لوحظ أنه عند تعريض عينه من محلول صبغة الملاكيت الخضراء في الهكســانون الحلقي لأشعة الضوء لفترة وجيزة ثم حفظ العينة في الظلام أن لون الصبغة الأخضر يختفي تدريجياً بمرور الزمن مما يدل على حدوث تفاعل ما بين الصبغة والمذيب (الهكسانون الحلقي)، لدراسة هذا التفاعل عملياً يلزم إجراء الآتي: أولا الطريقة المستخدمة في الدراسة : - استخدام الطرق الفيزيائية ( الطريقة الضوئية : قياس الامتصاصية مقابل الطول الموجي )
 لوحظ أنه عند تعريض عينه من محلول صبغة الملاكيت الخضراء في الهكســانون الحلقي لأشعة الضوء لفترة وجيزة ثم حفظ العينة في الظلام أن لون الصبغة الأخضر يختفي تدريجياً بمرور الزمن مما يدل على حدوث تفاعل ما بين الصبغة والمذيب (الهكسانون الحلقي)، لدراسة هذا التفاعل عملياً يلزم إجراء الآتي: أولا الطريقة المستخدمة في الدراسة : - استخدام الطرق الفيزيائية ( الطريقة الضوئية : قياس الامتصاصية مقابل الطول الموجي )")
80
1.تحديد الطول الموجي الذي تمتصه المواد المتفاعلة: تدرس العلاقة بين امتصاصية مخلوط المواد المتفاعلة مع التغير في الطول الموجي وذلك لتحديد طول موجي معين تجري عنده الدراسة قبل تعريض العينة للضوء (حدوث التفاعل) ويمثل الشكل الآتي العلاقة المذكورة حيث نجد أن المحلول يعطي أعلى امتصاصية عند الطول الموجي = 624 نانوميتر. شكل (12) العلاقة بين الطول الموجي والامتصاصية لمحلول صبغة الملاكيت الخضراء في الهكسانون الحلقي.
 العلاقة بين الطول الموجي والامتصاصية لمحلول صبغة الملاكيت الخضراء في الهكسانون الحلقي.")
81
. تحقيق قانون بير-لاميرت:
لكي يمكن استخدام امتصاصية المحلول مقياساً للتغيير في التركيز ىبد من التأكد من سريان قانون بير – لامبرت على المحاليل المستخدمة أي: A = εCl حيث A هي امتصاصية المحلول. و C هي التركيز المقابل للامتصاصية ε هي معامل الانطفاء وهو قيمة ثابتة للمحلول الواحد. l هي سمك المحلول المستخدم وهي ثابتة للخلية الواحدة . لذلك تحضر عدة محاليل قياسية معلومة التركيز وتقاس الامتصاصية المقابلة لكل تركيز، ثم ترسم العلاقة بين الامتصاصية المقاسه والتركيز المقابل لها ومن الشكل يحدد المدى من التركيز الذي ينطبق عليه قانون بير – لامبرت والذي يلزم ان تتم في الدراسة في مداه ويبين الشكل الآتي هذه العلاقة عند استخدام صبغة الملاكيت الخضراء في الهكسانون الحلقي. ويظهر الشكل أن تركيز العينات المستخدمة في الدراسة يلزم أن تنحصر بين (2.35 x x 3.3 x10-5) مولار والتي تقابل امتصاصية من ( ).
 مولار والتي تقابل امتصاصية. من ( ).")
82
3.تحديد الطول الموجي الذي تمتصه المواد الناتجة: بعد انتهاء التفاعل (أي اختفاء اللون تماماً) يدرس التغير في امتصاصية المحلول الناتج مقابل الطول الموجي لمعرفة هل يوجد طول موجي معين تمتصه المواد الناتجة من التفاعل أي الكشف عن قمة امتصاصية جديدة غير تلك التي تظهر للمواد المتفاعلة. وقد أظهرت الدراسة في حالة تفاعل صبغة الملاكيت الخضراء مع الهكسانون الحلقي عدم ظهور أي قمم امتصاصية جديدة لنواتج التفاعل.

83
القياسات التجريبية المستخدمة في الدراسة:
يتفاعل الهكسانون الحلقي مع صبغة الملاكيت الخضراء في وجود الضوء كعامل محفز ليعطي نواتج عديمة اللون حيث تعرض عينة من مخلوط التفاعل لفترة زمنية محددة لأشعة لمبة الزئبق ذات ضغط متوسط (تعطي أطوال موجبة محددة) ثم يدرس التفاعل بعيداً عن الضوء عن طريق قياس التغيير في تركيز المواد المتفاعلة في الظلام (بقياس امتصاصية المحلول) مقابل الزمن عند طول موجي = 624 نانوميتر ويطلق على هذا النوع من التفاعلات (تفاعلات مظلمة محفزة بالضوء) وقد تم إجراء نوعان من القياسات هما: -استخدام تركيز ابتدائي ثابت لصبغة الملاكيت الخضراء حيث تعرض العينات تحت الدراسة إلى الضوء لأزمنة مختلفة على التوالي وفي كل حالة تحفظ العينة بعد تعريضها للضوء في الظلام ويقاس التغير في الامتصاصية مقابل الزمن . - باستخدام تراكيز مختلفة من صبغة الملاكيت الخضراء وتعريض جميع العينات تحت الدراسة في هذه الحالة إلى الضوء لفترة زمنية محددة (حوالي 3 دقائق) تحفظ بعدها العينة في الظلام ويقاس التغيير في الامتصاصية مقابل الزمن.
 ثم يدرس التفاعل بعيداً عن الضوء عن طريق قياس التغيير في تركيز المواد المتفاعلة في الظلام (بقياس امتصاصية المحلول) مقابل الزمن عند طول موجي = 624 نانوميتر ويطلق على هذا النوع من التفاعلات (تفاعلات مظلمة محفزة بالضوء) وقد تم إجراء نوعان من القياسات هما: -استخدام تركيز ابتدائي ثابت لصبغة الملاكيت الخضراء حيث تعرض العينات تحت الدراسة إلى الضوء لأزمنة مختلفة على التوالي وفي كل حالة تحفظ العينة بعد تعريضها للضوء في الظلام ويقاس التغير في الامتصاصية مقابل الزمن . - باستخدام تراكيز مختلفة من صبغة الملاكيت الخضراء وتعريض جميع العينات تحت الدراسة في هذه الحالة إلى الضوء لفترة زمنية محددة (حوالي 3 دقائق) تحفظ بعدها العينة في الظلام ويقاس التغيير في الامتصاصية مقابل الزمن.")
84
كما أجريت تجارب لتحديد المادة التي تقوم بامتصاص الضوء وتكون المبادرة لحدوث التفاعل حيث عرضت عينات من الهكسانون الحلقي النقي (حوالي 3 مل) إلى الضوء لفترات زمنية مختلفة وفي كل حالة يضاف إلى المحلول المشعع حوالي 0.5 مل من محلول الصبغة المركز في الهكسانون الحلقي محفوظة في الظلام، ثم يقاس التغير في الامتصاصية للمحلول الناتج مقابل الزمن.
 إلى الضوء لفترات زمنية مختلفة وفي كل حالة يضاف إلى المحلول المشعع حوالي 0.5 مل من محلول الصبغة المركز في الهكسانون الحلقي محفوظة في الظلام، ثم يقاس التغير في الامتصاصية للمحلول الناتج مقابل الزمن.")
85
النتائج أعطت نتائج الامتصاصية مع الزمن خط إلى حد ما مستقيم عند رسم مقلوب الامتصاصية (1/A) مقابل الزمن في جميع القياسات السابقة أي أن التفاعل الناتج هو تفاعل من الدرجة الثانية . كذلك يستنتج من تماثل النتائج في حالة تعريض الصبغة مع المذيب إلى الضوء وعند تعريض المذيب فقط وإضافة الصبغة لاحقاً أن الذي يقوم بامتصاص الضوء والمبادر لحدوث التفاعل المظلم هو المذيب وليس الصبغة وعليه يمكن اقتراح الميكانيكية الآتية للتفاعل المحفز ضوئياً بين صبغة الملاكيت الخضراء والهكسانون الحلقي.
 مقابل الزمن في جميع القياسات السابقة أي أن التفاعل الناتج هو تفاعل من الدرجة الثانية . كذلك يستنتج من تماثل النتائج في حالة تعريض الصبغة مع المذيب إلى الضوء وعند تعريض المذيب فقط وإضافة الصبغة لاحقاً أن الذي يقوم بامتصاص الضوء والمبادر لحدوث التفاعل المظلم هو المذيب وليس الصبغة وعليه يمكن اقتراح الميكانيكية الآتية للتفاعل المحفز ضوئياً بين صبغة الملاكيت الخضراء والهكسانون الحلقي.")
86
يقوم الهكسانون الحلقي بإمتصاص الضوء أي: + hν → (1)
يقوم الهكسانون الحلقي بإمتصاص الضوء أي: + hν → (1) يتبع ذلك استخلاص لذرة هيدروجين من الوضع لجزئ مستقر من الهكسانون الحلقي لتكون الجذور الآتية: جذر الهكسانون الحلقي الذي في الخطوة (2) يكون مستقراً بسبب الاتزان الآتي: تتفاعل جذور الهكسانون الحلقي الحرة مع أيونات الصبغة الموجبة وتتكون جذور للصبغة:
 يتبع ذلك استخلاص لذرة هيدروجين من الوضع لجزئ مستقر من الهكسانون الحلقي لتكون الجذور الآتية: جذر الهكسانون الحلقي الذي في الخطوة (2) يكون مستقراً بسبب الاتزان الآتي: تتفاعل جذور الهكسانون الحلقي الحرة مع أيونات الصبغة الموجبة وتتكون جذور للصبغة:")
87
تتفاعل جذور الصبغة مع الأكسجين لتكوين جذور فوق أكسيد الصبغة وهذه الخطوة هي المحددة لسرعة التفاعل (تفاعل من الدرجة الثانية لا يظهر فيها المذيب كمادة متفاعلة ) : MG٠ + O بطئ MGOO٠ تفاعل جذور فوق الأكسيد مع الكيتون الحلقي وتستخلص منه ذرة هيدروجين ويتكون فوق هيدروكسيد الصبغة MGOO٠ → MGOOH + R٠ MGOOH → MGO٠ + OH٠ MGO٠ + → MGOH + ( R٠)
")
88
من الخطوة السابقة يظهر أن جذر الكيتون الذي يفترض تكوينه عند تعريض الهكسانون الحلقي إلى الضوء الذي تفاعل مع الصبغة يستمر تكوينه حتى في غياب الضوء من خلال الخطوات المفترضة سابقاً وهذا يفسر سبب استمرار التفاعل حتى في غياب الضوء. كذلك أمكن الاستدلال على وجود MGOH (وهي قاعدة عديمة اللون) بإضافة حمض HCl إلى ناتج التفاعل عديم اللون فيظهر لون الصبغة الأخضر ثانية مما يدل على حدوث التفاعل الآتي: MGOH + HCl → MGCl + H2O
 بإضافة حمض HCl إلى ناتج التفاعل عديم اللون فيظهر لون الصبغة الأخضر ثانية مما يدل على حدوث التفاعل الآتي: MGOH + HCl → MGCl + H2O .")
89
وتشير الرموز السابقة MGOH , MG+ إلى المركبات الآتية:
هيدروكسيد صبغة الملاكيت الخضراء أيون صبغة الملاكيت الخضراء (عديم اللون) (لون أخضر)
 (لون أخضر)")
90
الباب الثالث التفاعلات المعقدة 1- التفاعلات العكسية.
Reversible Reactions 2- التفاعلات المتتابعة. Consecutive Reactions 3- التفاعلات المتوازية. Parallel Reactions 4- التفاعلات السلسلية. Chain Reactions

91
- التفاعلات العكسية: جميع التفاعلات الكيميائية تقريباً من الناحية النظرية عكسية ولكن عادة بهمل التفاعل العكسي إذا لم يكن له تأثير ملحوظ في التركيز، وفي التفاعلات العكسية يسير التفاعل إلى حالة الاتزان أي أن التفاعل في اتجاه تكوين المواد الناتجة يعاكسه تفاعل آخر في اتجاه تكوين المواد المتفاعلة كما يتضح من المثال التالي: CH3COOH + C2H5OH → CH3OOC2H5 + H2O وعند دراسة حركة التفاعلات العكسية يجب أن يؤخذ في الاعتبار هذا المؤثر الجديد في سير التفاعل ومن أبسط التفاعلات العكسية الحالات التالية: أولاً: عندما يكون التفاعل في الاتجاهين من الدرجة الأولى. ثانياً: عندما يكون التفاعل في الاتجاه الأمامي من الدرجة الأولى، ويعاكسه تفاعل من الدرجة الثانية. ثالثاً: عندما يكون التفاعل في الاتجاهين من الدرجة الثانية.

92
تفاعلان من الدرجة الأولي يعاكس أحدهما الآخر
الحالة (أ) : عندما يكون تركيز المواد الناتجة (B) صفراً عند بداية التفاعل ثم بمرور الوقت يكون تركيز المواد الناتجة (B) هو ما تفاعل من المادة (A) فإذا أفترض حدوث التفاعل الآتي: A 𝐾1 𝐾− B على K-1, K1 حيث ثوابت سرعة التفاعل النوعي بالنسبة إلى التفاعل في الاتجاه الأمامي والاتجاه المعاكس له هي التوالي. عند بداية التفاعل يكون التركيز الابتدائي للمادة (A) هو a وبعد مرور وقت على التفاعل يصبح تركيز المادة هو (a-x) بينما تركيز المادة (B) هو x، وعلى هذا تكتب معادلة سرعة التفاعل كالتالي: dx / dt = K1 (a – x) – K-1 X حيث أن المادة (B) تكونت نتيجة تفاعل المادة (A) في الاتجاه الأمامي ولكن بعد فترة زمنية معينة من بداية التفاعل، تنقص المادة (B) بمرور الوقت نتيجة التفاعل العكسي وبذلك يصل التفاعل إلى حالة الاتزان. بفرض أن Xe تمثل تركيز المادة (B) عند الاتزان فإن تركيز المادة (A) في حالة الاتزان يكون (a-xe) ومحصلة التغيير عند نقطة الاتزان تساوي صفراً، أي :
 : عندما يكون تركيز المواد الناتجة (B) صفراً عند بداية التفاعل ثم بمرور الوقت يكون تركيز المواد الناتجة (B) هو ما تفاعل من المادة (A) فإذا أفترض حدوث التفاعل الآتي: A 𝐾1 𝐾−1 B. على K-1, K1 حيث ثوابت سرعة التفاعل النوعي بالنسبة إلى التفاعل في الاتجاه الأمامي والاتجاه المعاكس له هي التوالي. عند بداية التفاعل يكون التركيز الابتدائي للمادة (A) هو a وبعد مرور وقت على التفاعل يصبح تركيز المادة هو (a-x) بينما تركيز المادة (B) هو x، وعلى هذا تكتب معادلة سرعة التفاعل كالتالي: dx / dt = K1 (a – x) – K-1 X. حيث أن المادة (B) تكونت نتيجة تفاعل المادة (A) في الاتجاه الأمامي ولكن بعد فترة زمنية معينة من بداية التفاعل، تنقص المادة (B) بمرور الوقت نتيجة التفاعل العكسي وبذلك يصل التفاعل إلى حالة الاتزان. بفرض أن Xe تمثل تركيز المادة (B) عند الاتزان فإن تركيز المادة (A) في حالة الاتزان يكون (a-xe) ومحصلة التغيير عند نقطة الاتزان تساوي صفراً، أي :")
93
وبالتعويض عن قيمة K-1 في معادلة السرعة ( معادلة 33) ينتج أن:
dx/dt = K1 (a – Xe ) – K-1 Xe = (34) K-1 = K1 (a – Xe ) / Xe (35) وبالتعويض عن قيمة K-1 في معادلة السرعة ( معادلة 33) ينتج أن: dx / dt = K1 (a – X ) – K-1(a – Xe) / Xe .X dx / dt = [K1 a/Xe] (Xe – X ) بتكامل المعادلة مع الأخذ في الاعتبار أن X = صفر عند بداية التفاعل أي عندما t = 0 يتم الحصول على : K1 = (Xe / a t) ln [Xe /( Xe – X)] (36) فإذا عرفت قيمة Xe أمكن حساب قيمة K1 من قياس (X) في فترات متتالية . وأحياناً لغرض الموازنة توضع معادلة ثابت سرعة التفاعل في شكل آخر وعلى هذا فان المعادلة (35) يمكن أن توضع على الصورة التالية: K-1 .Xe = K1 ( a – Xe) = K1a – K1. Xe
 – K-1 Xe = 0 (34) K-1 = K1 (a – Xe ) / Xe (35) وبالتعويض عن قيمة K-1 في معادلة السرعة ( معادلة 33) ينتج أن: dx / dt = K1 (a – X ) – K-1(a – Xe) / Xe .X. dx / dt = [K1 a/Xe] (Xe – X ) بتكامل المعادلة مع الأخذ في الاعتبار أن X = صفر عند بداية التفاعل أي عندما t = 0 يتم الحصول على : K1 = (Xe / a t) ln [Xe /( Xe – X)] (36) فإذا عرفت قيمة Xe أمكن حساب قيمة K1 من قياس (X) في فترات متتالية . وأحياناً لغرض الموازنة توضع معادلة ثابت سرعة التفاعل في شكل آخر وعلى هذا فان المعادلة (35) يمكن أن توضع على الصورة التالية: K-1 .Xe = K1 ( a – Xe) = K1a – K1. Xe.")
94
( K-1 + K1) Xe = K1. A Xe / a = K1 / (K-1 + K 1 ) بالتعويض عن قيمة Xe / a في المعادلة (36) ينتج أن : ] K1 = (K1 / K1 + K-1 ) / t ln [ Xe / ( Xe – X) Or K1 + K-1 = 1/t ln [Xe / ( Xe – X ) ] (37) بمقارنة هذه المعادلة بمعادلة الدرجة الأولى وصيغتها : K = 1/ t ln a / ( a-x ) يتضح التشابه التالي : التركيز في حالة الاتزان Xe حل محل التركيز الابتدائي a في معادلة الدرجة الأولى. مجموع ثابتي سرعة التفاعل في الاتجاهين ( K1 + K-1 ) حلت محل ثابت سرعة التفاعل النوعي K.
 بالتعويض عن قيمة Xe / a في المعادلة (36) ينتج أن : ] K1 = (K1 / K1 + K-1 ) / t ln [ Xe / ( Xe – X) Or K1 + K-1 = 1/t ln [Xe / ( Xe – X ) ] (37) بمقارنة هذه المعادلة بمعادلة الدرجة الأولى وصيغتها : K = 1/ t ln a / ( a-x ) يتضح التشابه التالي : التركيز في حالة الاتزان Xe حل محل التركيز الابتدائي a في معادلة الدرجة الأولى. مجموع ثابتي سرعة التفاعل في الاتجاهين ( K1 + K-1 ) حلت محل ثابت سرعة التفاعل النوعي K.")
95
الحالة (ب) : عندما يكـــون الـــــتركيز الابـــــتدائي للمادة (A) هـو (a) ، والتركيز الابتدائي للمادة (B) هو (b) . بفرض التفاعل التالي: A B فإن معدل السرعة تكتب كالتالي : dx / dt = K1 (a –x ) – K-1 (b + x) dx / dt = K1 a – K1 X – K-1 – K-1 X dx / dt = ( K1 a – K-1 b ) – X ( K1 + K-1 ) (38) ولكن عند الاتزان : dx/dt = 0 , X = Xe التركيز عند الاتزان: :. Xe = (K1 a – K-1 b) / (K1 +K-1 ) بفرض أن : 1. K1 a – K-1b = A 2. (– K1 – K-1 ) = B
 – K-1 (b + x) dx / dt = K1 a – K1 X – K-1 – K-1 X dx / dt = ( K1 a – K-1 b ) – X ( K1 + K-1 ) (38) ولكن عند الاتزان : dx/dt = 0 , X = Xe التركيز عند الاتزان: :. Xe = (K1 a – K-1 b) / (K1 +K-1 ) بفرض أن : 1. K1 a – K-1b = A 2. (– K1 – K-1 ) = B .")
96
بالتعويض عن قيمة Xe Xe = -A / B (39) كذلك بالتعويض في المعادلة (38) ينتج أن : dx / dt = A + BX بفصل المتغيرات: dx / (A + BX ) = dt (40) بتكامل المعادلة (40) ينتج أن: ( 1/B) ln ( A + BX) = t + K΄ (41) ولتعيين قيمة K΄ ثابت التكامل نفرض الحالة t = 0 أي عند x = 0 بالتعويض في المعادلة (41) ينتج أن : K΄ = (1/B ) ln A
 كذلك بالتعويض في المعادلة (38) ينتج أن : dx / dt = A + BX بفصل المتغيرات: dx / (A + BX ) = dt (40) بتكامل المعادلة (40) ينتج أن: ( 1/B) ln ( A + BX) = t + K΄ (41) ولتعيين قيمة K΄ ثابت التكامل نفرض الحالة t = 0 أي عند x = 0 بالتعويض في المعادلة (41) ينتج أن : K΄ = (1/B ) ln A")
97
وبالتعويض في المعادلة (41) ينتج أن :
(1 / B ) ln (A + BX) = t + (1 / B ) ln A (1 / B ) ln (A + BX) – (1 / B) ln A = t بالتعويض عن قيمة (1 / B) في المعادلة السابقة ينتج أن : ( -1 / K1 + K-1 ) ln ( A + BX ) + (1 / K1 + K-1 ) ln A = t (1 / t) ln A/ ( A + BX) = K1 + K-1 بقسمة البسط والمقام في الطرف الأيسر على المقدار B ينتج أن : ( 1 / t) ln A/B /(A/B + B/B X) = K1 + K-1 بالتعويض عن قيمة A/B (39) ينتج أن : 1/t ln [ - Xe/ (-Xe +X) ] = K1 + K (42) 1/t ln (+ Xe/ (Xe – X) ) = K1 + K-1 والمعادلة (42) لها نفس شكل المعادلة (37) التي تمثل الحالة (أ) ولكن الاختلاف كان في قيمة التركيز Xe في حالة الاتزان.
 ln (A + BX) = t + (1 / B ) ln A. (1 / B ) ln (A + BX) – (1 / B) ln A = t. بالتعويض عن قيمة (1 / B) في المعادلة السابقة ينتج أن : ( -1 / K1 + K-1 ) ln ( A + BX ) + (1 / K1 + K-1 ) ln A = t. (1 / t) ln A/ ( A + BX) = K1 + K-1. بقسمة البسط والمقام في الطرف الأيسر على المقدار B ينتج أن : ( 1 / t) ln A/B /(A/B + B/B X) = K1 + K-1. بالتعويض عن قيمة A/B (39) ينتج أن : 1/t ln [ - Xe/ (-Xe +X) ] = K1 + K-1 (42) 1/t ln (+ Xe/ (Xe – X) ) = K1 + K-1. والمعادلة (42) لها نفس شكل المعادلة (37) التي تمثل الحالة (أ) ولكن الاختلاف كان في قيمة التركيز Xe في حالة الاتزان.")
98
أمثلة على التفاعلات العكسية
1-تحويل جاما – هيدروكسي حمض البيوتيرك إلي اللاكتون : هذا التفاعل على التفاعل العكسي من الدرجة الأولى في الاتجاهين، لذلك فالتركيز الابتدائي لمادة اللاكتون يساوي صفراً عند بداية التفاعل، ثم يزداد تركيزه حتى يصل إلى حالة الاتزان، ويكون تركيزه في هذه الحالة Xe = وزن جزئ في اللتر، كما أن قيمة K-1, K1 يمكن حسابها معرفة (X) في الفترات المتتالية وبالاستعانة بمعادلة ثابت السرعة النوعي (المعادلة (37)). K1 + K-1 = 1 / t ln (Xe / ( Xe – X)) وجد أن قيمة K-1, K1 = 1.56 × 10-4 ثانية-1، وهذه القيمة في حدود الخطأ التجريبي لثابت سرعة التفاعل النوعي K-1, K1، تؤكد أن درجة التفاعل هي الدرجة الأولى، كما أنه بمعرفة قيمة التركيز الابتدائي عند التوازن يمكن إيجاد كل من قيمة K-1, K1 على حدة.
 في الفترات المتتالية وبالاستعانة بمعادلة ثابت السرعة النوعي (المعادلة (37)). K1 + K-1 = 1 / t ln (Xe / ( Xe – X)) وجد أن قيمة K-1, K1 = 1.56 × 10-4 ثانية-1، وهذه القيمة في حدود الخطأ التجريبي لثابت سرعة التفاعل النوعي K-1, K1، تؤكد أن درجة التفاعل هي الدرجة الأولى، كما أنه بمعرفة قيمة التركيز الابتدائي عند التوازن يمكن إيجاد كل من قيمة K-1, K1 على حدة.")
99
2- تحويل الفا – جلوكوز إلي بيتا جلوكوز: الفا – جلوكوز بيتا جلوكوز هذا التفاعل مثال على التفاعل العكسي من الدرجة الأولى في اتجاهين عند تحضير محلول حديث يحتوي على الفا – جلوكوز النقي في الماء يكون تركيز البيتا – جلوكوز يساوي صفراً، ثم يزداد تركيزها حتى يصل إلى حالة الاتزان. هذا التفاعل يمكن متابعته باستخدام جهاز قياس الاستقطاب.

100
التفاعل بين الهيدورجين واليود:
هذا التفاعل بسيط من وجهة نظر حركة التفاعلات الكيميائية، وبعد مثالاً جيداً على التفاعلات العكسية من الدرجة الثانية. ويتم هذا التفاعل في درجة الحرارة عالية نسبياً بين ºم مئوية وسرعة سير التفاعل يمكن قياسها. وتتلخص الطريقة في أن يوضع الهيدروجين واليود في زجاجة دائرية، ثم يلحم عليها وبعد تسخين الزجاجة تبرد فجائياً ويتم تحليل محتواها، ولأنه ليس من الممكن أخذ كميات متساوية من الهيدروجين واليود عند بداية التفاعل فإن تركيزها الابتدائي سيكون مختلفاً، أما عن ميكانيكية التفاعل فهو تفاعل عكسي من الدرجة الثانية في الاتجاهين ويمكن وضع معادلة التفاعل كالتالي: H2 + I K1 K− HI ومحصلة معدل التفاعل لتكوين يوديد الهيدروجين هي سرعة التفاعل في الاتجاه الأمامي ناقصاً سرعة التفاعل في الاتجاه العكسي وعلى هذا يمكن وضع سرعة التفاعل كالتالي: dCHl / dt = K1 CH2 .Cl2 –K-1 C2Hl بمعرفة التركيز الابتدائي للهيدروجين واليود، ومن نتائج التحليل يمكن معرفة تركيز يوديد الهيدروجين (X) بعد مرور زمن قدره t كما يمكن إيجاد قيمة ثابت التفاعل النوعي في أي إتجاه K1 أو K-1 .
 بعد مرور زمن قدره t كما يمكن إيجاد قيمة ثابت التفاعل النوعي في أي إتجاه K1 أو K-1 .")
101
مسائل على التفاعلات العكسية
يعطي الحامض هيدروكسي بيوترك في المحلول المائي بيوترولاكتون حسب المعادلة : CH2OH.CH2.COOH → CH2. CH2. CH2.CO +H2O │……..O │ قد أعطيت قيم اعتماد تركيز حامض هيدروكسي بيوترك على الزمن عند 25م كالآتي : C (mole. L-1) t (min) 10.25 100 18.23 6.67 220 15.84 21 4.96 ∞ 13.25 50 أوجدي ثوابت المعدل للتفاعلات في كلا الاتجاهين وثابت التوازن للتفاعل الأمامي، أفترض أن التفاعل العكسي في المحلول المائي شبه أحادي الجزيئات – pseudo Unimolecular .
 t (min) ∞ أوجدي ثوابت المعدل للتفاعلات في كلا الاتجاهين وثابت التوازن للتفاعل الأمامي، أفترض أن التفاعل العكسي في المحلول المائي شبه أحادي الجزيئات – pseudo Unimolecular .")
102
CH3COOH + C2H5OH K CH3COOHC2H5+H2O K-1
- عند دراسة تكون خلات الأيثيل من حمض الخليك والكحول الإيثيلي باستخدام تركيز ثابت من حمض HCI كعامل محفز للتفاعل وذلك حسب المعادلة : CH3COOH + C2H5OH K CH3COOHC2H5+H2O K-1 عوير 1 مل في كل مرة من مخلوط التفاعل باستخدام ع من القاعدة المستخدمة ويبين الجدول الآتي النتائج التي حصل عليها أثناء الدراسة: بمعرفة أن مرتبة التفاعل للمواد السابقة المتفاعلة والناتجة هو الدرجة الأولى. K-1, K1 أحسبي كلاً من حجم القاعدة المأخوذ الزمن 18.29 148 24.37 15.15 313 22.2 44 14.5 384 21.53 62 14.09 442 19.5 108 12.68 ∞ 19.26 117 فإذا كان التركيز الابتدائي لكل من : CH3COOH = 1M H2O =12.756M C2H5OH=12.75M CH3COOC2H5=0

103
.التفاعلات المتتابعة كثيراً ما يحدث أن يصبح ناتج ما هو نفسه مادة متفاعلة لتفاعل لاحق، وقد تكون هناك مجموعة من الخطوات المتتابعة أى أن نواتج التفاعل يتفاعل بعضها مع بعض ليعطي نواتج أخرى وهكذا .. وقد أمكن في أبسط الحالات فقط إيجاد حل للمعادلات التفاضلية التي تمثل هذا النوع من التفاعل، والتفاعلات المتتابعة مهمة جداً ولاسيما في عمليات البلمرة Polymerization . ومن ابسط الأمثلة على هذا النوع من التفاعلات أن تكون التفاعلات المتعاقبة من الدرجة الأولى مع عدم وجود تفاعلات عكسية وعليه يمكن وضع معادلة التفاعل كما يلي A K1 B K2 D حيث K1 ، K2 هي ثوابت التفاعل النوعي ، فإذا كانت CD, CB, CA تمثل التركيز للمواد D, B, A على التوالي فإن معادلة سرعة التفاعل بالنسبة للمادة المتفاعلة A هي - d CA / dt = K1 CA = K1 [A]

104
وبتكامل المعادلة ينتج : K1 = 1/t ln (a/ Ca Or K1
وبتكامل المعادلة ينتج : K1 = 1/t ln (a/ Ca Or K1. t = ln a – ln CA بأخذ لن الطرفين: CA = a. ē K1t (46) ومعادلة سرعة التفاعل لتكون المادة D هي : - dC / dt = K2 CB = K2 [B] (47) محصلة سرعة التفاعل لتكوين المادة B عبارة عن سرعة التفاعل لتكوينها من المادة A ناقصاً سرعة تفاعلها لتعطي المادةD وهي كالتالي: d [B] / dt = K1 . CA – K2 CB (48) بالتعويض عن قيمة CA من المعادلة (46) ينتج أن : d [B] / dt = K1ae –K1t – K2 CB
 ومعادلة سرعة التفاعل لتكون المادة D هي : - dC / dt = K2 CB = K2 [B] (47) محصلة سرعة التفاعل لتكوين المادة B عبارة عن سرعة التفاعل لتكوينها من المادة A ناقصاً سرعة تفاعلها لتعطي المادةD وهي كالتالي: d [B] / dt = K1 . CA – K2 CB (48) بالتعويض عن قيمة CA من المعادلة (46) ينتج أن : d [B] / dt = K1ae –K1t – K2 CB")
105
بضرب طرفي المعادلة السابقة في القيمة e k2t ينتج أن: d [B] / dt
بضرب طرفي المعادلة السابقة في القيمة e k2t ينتج أن: d [B] / dt .e K2t = K1ae (K2- K1)t – K2 CB. e K2t :.d [B] / dt. e K2t + K2 CB. eK2 = K1 a e (K2 - K1)t حاصل ضرب دالتين ، وعليه : d / dt (e K2t . CB) = K1 a e (K2 - K1)t بتكامل المعادلة السابقة ينتج أن : ∫d( e K2t. CB ) = ∫ K1ae (K2 – K1) t dt :. e K2t. CB = K1 / K2 – K1 ae (K2 – K1) t + K΄ لإيجاد قيمة ثابت التكامل K΄ نقسم طرفي المعادلة e K2t وبذلك نحصل على : CB = K1a / ( K2 – K1) ē K1t + K΄ ē K2t (49)
![بضرب طرفي المعادلة السابقة في القيمة e k2t ينتج أن: d [B] / dt](http://slideplayer.ae/slide/15551337/88/images/105/%D8%A8%D8%B6%D8%B1%D8%A8+%D8%B7%D8%B1%D9%81%D9%8A+%D8%A7%D9%84%D9%85%D8%B9%D8%A7%D8%AF%D9%84%D8%A9+%D8%A7%D9%84%D8%B3%D8%A7%D8%A8%D9%82%D8%A9+%D9%81%D9%8A+%D8%A7%D9%84%D9%82%D9%8A%D9%85%D8%A9+e+k2t+%D9%8A%D9%86%D8%AA%D8%AC+%D8%A3%D9%86%3A+d+%5BB%5D+%2F+dt.jpg "بضرب طرفي المعادلة السابقة في القيمة e k2t ينتج أن: d [B] / dt .e K2t = K1ae (K2- K1)t – K2 CB. e K2t :.d [B] / dt. e K2t + K2 CB. eK2 = K1 a e (K2 - K1)t حاصل ضرب دالتين ، وعليه : d / dt (e K2t . CB) = K1 a e (K2 - K1)t بتكامل المعادلة السابقة ينتج أن : ∫d( e K2t. CB ) = ∫ K1ae (K2 – K1) t dt :. e K2t. CB = K1 / K2 – K1 ae (K2 – K1) t + K΄ لإيجاد قيمة ثابت التكامل K΄ نقسم طرفي المعادلة e K2t وبذلك نحصل على : CB = K1a / ( K2 – K1) ē K1t + K΄ ē K2t (49)")
106
عند بداية التفاعل أى عند t = صفر ، فإن CB = 0 :
عند بداية التفاعل أى عند t = صفر ، فإن CB = 0 :. بالتعويض في المعادلة (49) ينتج أن: CB = K1a / ( K2 – K1 ) .e –K1xo + Kَ e-K2 xo = o :. Kَ = - K1a / ( K2 – K-1 ) بالتعويض K΄ في المعادلة (49) بقيمتها نجد أن : CB = K1a / ( K2 – K-1 ) e-K1t - K1a / ( K2 – K-1 ) e-K2t CB = K1a / ( K2 – K-1 ) (e-K1t - e-K2t ) (50) فإن عرفت قيمة K2 , K1 أصبح من السهل إيجاد قيمة CA , CB , CD بعد مرور زمن = t على التفاعل، حيث يمكن تعيين قيمة CA من المعادلة (46) وتركيز المادة CB من المعادلة (50) ، أما قيمة CD فيمكن حسابها بمعرفة التركيز الابتدائي a من العلاقة : a = CA + CB + CD عند دراسة العلاقة بين تركيز كل من [A] , [B] , [D] مع الزمن تحصل على الشكل المبين:
 ينتج أن: CB = K1a / ( K2 – K1 ) .e –K1xo + Kَ e-K2 xo = o :. Kَ = - K1a / ( K2 – K-1 ) بالتعويض K΄ في المعادلة (49) بقيمتها نجد أن : CB = K1a / ( K2 – K-1 ) e-K1t - K1a / ( K2 – K-1 ) e-K2t CB = K1a / ( K2 – K-1 ) (e-K1t - e-K2t ) (50) فإن عرفت قيمة K2 , K1 أصبح من السهل إيجاد قيمة CA , CB , CD بعد مرور زمن = t على التفاعل، حيث يمكن تعيين قيمة CA من المعادلة (46) وتركيز المادة CB من المعادلة (50) ، أما قيمة CD فيمكن حسابها بمعرفة التركيز الابتدائي a من العلاقة : a = CA + CB + CD عند دراسة العلاقة بين تركيز كل من [A] , [B] , [D] مع الزمن تحصل على الشكل المبين:")
107
يزداد حتى يصل إلى القمة ثم ينقص بينما التركيز
والشكل (3) يوضح أن [B] يزداد حتى يصل إلى القمة ثم ينقص بينما التركيز [A] ينقص طوال فترة التفاعل، والتركيز [D] يزداد باستمرار حتى نهاية التفاعل. الــــزمــــن ومن الأمثلة على هذا النوع من التفاعل تجزؤ الأستيون بتأثير الحرارة إلى الميثان والمركب المتوسط الكيتين الذي يتجزأ بدوره إلى اكسيد الكربون والايثلين ويمكن وضع معادلات التفاعل على النحو التالي: ( CH3 )2 C = O → CH4 + CH2 = C = O → [ 1/2 C2 H4 + C = O] كيتين تركيز المركب المتوسط (الكيتين) يزداد حتى يصل إلى القمة ثم ينقص، وفي الحقيقة إن تجزأ الأستيون بتأثير الحرارة أكثر تعقيداً مما تشير إليه المعادلات.
 يوضح أن [B] يزداد حتى يصل إلى القمة ثم ينقص بينما التركيز. [A] ينقص طوال فترة التفاعل، والتركيز [D] يزداد باستمرار حتى نهاية التفاعل. الــــزمــــن. ومن الأمثلة على هذا النوع من التفاعل تجزؤ الأستيون بتأثير الحرارة إلى الميثان والمركب المتوسط الكيتين الذي يتجزأ بدوره إلى اكسيد الكربون والايثلين ويمكن وضع معادلات التفاعل على النحو التالي: ( CH3 )2 C = O → CH4 + CH2 = C = O → [ 1/2 C2 H4 + C = O] كيتين. تركيز المركب المتوسط (الكيتين) يزداد حتى يصل إلى القمة ثم ينقص، وفي الحقيقة إن تجزأ الأستيون بتأثير الحرارة أكثر تعقيداً مما تشير إليه المعادلات.")
108
كذلك من الأمثلة على هذا النوع ما قام به العالمان (هاركورت وأيسون) من دراسة التفاعل بين برمنجنات البوتاسيوم وحمض الأكساليك وكبريتات المنجنوز في وجود حمض الكبريتيك، حيث يتم التفاعل وفق الآتي: يتكون ثاني اكسيد المنجنوز (MnO2) بسرعة ثم يتبعه تفاعل آخر بطئ فيتاكسد بواسطة برمنجنات البوتاسيوم ويتكون مركب وسطي ربما يكون سابع اكسيد المنجنيز Mn2 O7. يلحق به تفاعل آخر هو إختزال المركب المتوسط حمض الأكساليك إلى أول اكسيد المنجنوز MnO . إن كلاً من برمنجنات البوتاسيوم وحمض الأكساليك موجود في زيادة كبيرة ولا يتأثر تركيز كل منهما بمرور التفاعل من ذلك نجد أن سرعة التفاعل ودرجته تعتمد على تركيز ثاني اكسيد المنجنيز MnO2 ويمكن كتابة معادلة التفاعل كالآتي: MnO2 → K1 Mn2 O7 →K2 MnO كذلك يمكن متابعة سير التفاعل بإضافة يوديد البوتاسيوم على فترات متتالية ثم معايرة اليود المنطلق بواسطة محلول قياسي من ثيوكبريتات الصوديوم .. ومن هذا يمكن معرفة قيمة التراكيز المختلفة الموجودة في هذا التفاعل.

109
مسائل 1- تفاعلات متتابعان من المرتبة الأولى A K1 B K2 C لهما ثابتي K1 ، K2سرعة حددي أي ثابتي السرعتين يقرر معدل تكوين المادة C إذا كان : a) K1 » K2 b) K1 « K2
 K1 » K2. b) K1 « K2.")
110
التفاعلات المتوازية تحدث التفاعلات المتوازية أو الجانبية عندما تتفكك مادة أو تتفاعل بأكثر من طريقة مما يؤدي إلى تكوين نوعين أو أكثر من المواد الناتجة وهذه تشمل تفاعلات الإحلال التى تؤدي إلى تكوين عدد من المركبات المتشابهة في التركيب الكيميائي مع اختلاف في الخواص والتى تعرف باسم المماثلات أو المتشاكلات isomers فعند نترجة كلوروبنزين يتم الحصول على ثلاثة أنواع من نيتروكلوروبنزين وكل واحد منها له سرعة تكوين تختلف عن الأخرى وهذه الأنواع هي : أورثو – ميتا – بارانيتروكلوروبنزين. كذلك الكحولات من الممكن أن تفقد جزيئاً من المادة وتعطي الأوليفينات أو تفقد جزئاً من الهيدروجين وتعطي الالدهيدات كما في المعادلة الآتية:

111
وبالاختيار المناسب للحافز المنشط ولدرجة الحرارة يمكن أن تكون سرعة أحد المكونات أسرع من الآخر، وفي هذه الحالة تعتمد كمية الناتج لكل منهما على سرعة التفاعل النسبية ، وهذه إحدى مميزات دراسة حركية التفاعلات الكيميائية حيث تساعد على معرفة أي من النواتج سيكون الغالب، وبذلك يمكن توجيه التفاعل للحصول على نواتج معينة ، وفي هذه الحالة التي يتغلب فيها أحد التفاعلات على التفاعلات الأخرى التى تكون اكثر بطئاً، فإنه يصبح هو الناتج الغالب تسمى هذه بالتفاعلات المتنافسة (Competing Reactions) وهذه كثيرة الحدوث ولا سيما في الكيمياء العضوية. ومن أبسط حالات التفاعلات المتوازية أن يكون التفاعلات من الدرجة الأولى، ويمكن وضع معادلة التفاعل على النحو التالي : ولإيجاد معادلة سرعة التفاعل نفترض أن ليس هناك أي تفاعلات عكسية فإذا كان التركيز الابتدائي للمادة نجد أن : A هو a

112
dx / dt = K1 ( a – x ) + K2 ( a – x ) dx / dt = ( K1 + K2 ) ( a – x ) بفصل المتغيرات في المعادلة السابقة : dx / ( a – X ) = ( K1 + K2 ) dt (51) بتكامل المعادلة (51) نحصل على : - ln ( a – x ) = ( K1 + K2 ) t + Kَ ( 52) بالتعويض في المعادلة (52) تعيين قيمة K΄ ينتج أن : Kَ = - ln a بالتعويض عن قيمة K΄ في المعادلة (52) ينتج أن : lna – ln ( a – x) = ( K1 + K2 ) t K1 + K2 = 1 / t ln a / ( a – x) (53)
 + K2 ( a – x ) dx / dt = ( K1 + K2 ) ( a – x ) بفصل المتغيرات في المعادلة السابقة : dx / ( a – X ) = ( K1 + K2 ) dt (51) بتكامل المعادلة (51) نحصل على : - ln ( a – x ) = ( K1 + K2 ) t + Kَ ( 52) بالتعويض في المعادلة (52) تعيين قيمة K΄ ينتج أن : Kَ = - ln a بالتعويض عن قيمة K΄ في المعادلة (52) ينتج أن : lna – ln ( a – x) = ( K1 + K2 ) t K1 + K2 = 1 / t ln a / ( a – x) (53)")
113
ولما كان معدل تكوين المادة B هو : d [B] / dt = [B] = K1 ( a – x ) ومعدل تكوين المادة D هو : d [D] / dt = [D] = K2 ( a – x ) d [B] / d [D] = K1 / K2 = [B] / [D] ثابت = تركيز المادة (54) أي أن النسبة بين التركيز المادةB إلى تركيز المادة D في أي لحظة يساوي مقدار ثابت بشرط أن تكون درجة التفاعل واحدة بالنسبة للمواد الناتجة .
![ولما كان معدل تكوين المادة B هو : d [B] / dt = [B] = K1 ( a – x ) ومعدل تكوين المادة D هو : d [D] / dt = [D] = K2 ( a – x ) d [B] / d [D] = K1 / K2 = [B] / [D] ثابت = تركيز المادة (54) أي أن النسبة بين التركيز المادةB إلى تركيز المادة D في أي لحظة يساوي مقدار ثابت بشرط أن تكون درجة التفاعل واحدة بالنسبة للمواد الناتجة .](http://slideplayer.ae/slide/15551337/88/images/113/%D9%88%D9%84%D9%85%D8%A7+%D9%83%D8%A7%D9%86+%D9%85%D8%B9%D8%AF%D9%84+%D8%AA%D9%83%D9%88%D9%8A%D9%86+%D8%A7%D9%84%D9%85%D8%A7%D8%AF%D8%A9+B+%D9%87%D9%88+%3A+d+%5BB%5D+%2F+dt+%3D+%5BB%5D+%3D+K1+%28+a+%E2%80%93+x+%29+%D9%88%D9%85%D8%B9%D8%AF%D9%84+%D8%AA%D9%83%D9%88%D9%8A%D9%86+%D8%A7%D9%84%D9%85%D8%A7%D8%AF%D8%A9+D+%D9%87%D9%88+%3A+d+%5BD%5D+%2F+dt+%3D+%5BD%5D+%3D+K2+%28+a+%E2%80%93+x+%29+d+%5BB%5D+%2F+d+%5BD%5D+%3D+K1+%2F+K2+%3D+%5BB%5D+%2F+%5BD%5D+%D8%AB%D8%A7%D8%A8%D8%AA+%3D+%D8%AA%D8%B1%D9%83%D9%8A%D8%B2+%D8%A7%D9%84%D9%85%D8%A7%D8%AF%D8%A9+%2854%29+%D8%A3%D9%8A+%D8%A3%D9%86+%D8%A7%D9%84%D9%86%D8%B3%D8%A8%D8%A9+%D8%A8%D9%8A%D9%86+%D8%A7%D9%84%D8%AA%D8%B1%D9%83%D9%8A%D8%B2+%D8%A7%D9%84%D9%85%D8%A7%D8%AF%D8%A9B+%D8%A5%D9%84%D9%89+%D8%AA%D8%B1%D9%83%D9%8A%D8%B2+%D8%A7%D9%84%D9%85%D8%A7%D8%AF%D8%A9+D+%D9%81%D9%8A+%D8%A3%D9%8A+%D9%84%D8%AD%D8%B8%D8%A9+%D9%8A%D8%B3%D8%A7%D9%88%D9%8A+%D9%85%D9%82%D8%AF%D8%A7%D8%B1+%D8%AB%D8%A7%D8%A8%D8%AA+%D8%A8%D8%B4%D8%B1%D8%B7+%D8%A3%D9%86+%D8%AA%D9%83%D9%88%D9%86+%D8%AF%D8%B1%D8%AC%D8%A9+%D8%A7%D9%84%D8%AA%D9%81%D8%A7%D8%B9%D9%84+%D9%88%D8%A7%D8%AD%D8%AF%D8%A9+%D8%A8%D8%A7%D9%84%D9%86%D8%B3%D8%A8%D8%A9+%D9%84%D9%84%D9%85%D9%88%D8%A7%D8%AF+%D8%A7%D9%84%D9%86%D8%A7%D8%AA%D8%AC%D8%A9+..jpg "ولما كان معدل تكوين المادة B هو : d [B] / dt = [B] = K1 ( a – x ) ومعدل تكوين المادة D هو : d [D] / dt = [D] = K2 ( a – x ) d [B] / d [D] = K1 / K2 = [B] / [D] ثابت = تركيز المادة (54) أي أن النسبة بين التركيز المادةB إلى تركيز المادة D في أي لحظة يساوي مقدار ثابت بشرط أن تكون درجة التفاعل واحدة بالنسبة للمواد الناتجة .")
114
عند درجات الحرارة العالية ( ºم) يتفكك حامض الخليك إلى ثاني أكسيد الكربون والميثان وفي الوقت نفسه إلى الكيتين والماء كما يلي : وقد أعطيت قيمة ثابت سرعة التفاعل للتفاعل الأول (K1) = 3.74 ثانية-1 وبالنسبة لثابت سرعة التفاعل للتفاعل الثاني (K2) = 4.65 ثانية-1 . احسبي : الزمن اللازم لتفاعل 99% من حامض الخليك . الناتج الأقصى للكيتين (بالنسبة المئوية لحامض الخليك الموجود في الأصل) الذي يمكن التوصل له عند الدرجة الحرارية المعطاة.
 الذي يمكن التوصل له عند الدرجة الحرارية المعطاة.")
115
حجم هيدروكسيد الباريوم المأخوذ
يتحول حمض ثنائي كمفور 3- كربوكسيل عند درجات الحرارة العالية إلى الكامفور في وجود الكحول الايثيلي كمذيب ويتحرر نتيجة لذلك CO2 حسب المعادلة : C10H15OCOOH K C10H16O+CO2 وبدراسة حدوث هذا التفاعل في الأوساط الأخرى ، ثبت حدوث تفاعل جانبي هو : C10H15OCOOH + C2H5OH K2 C10H15OCOOC2H5 + H2O وكانت النتائج التي حصل عليها عند تفكك هذا الحمض في الكحول كالآتي : CO2 (gr) حجم هيدروكسيد الباريوم المأخوذ t ( min ) -- 20.00 0,0841 16.26 10 0,1545 13.25 20 0,2095 10.68 30 0.2482 8.74 40 0.3045 5.88 60 0.3556 3.99 80 حيث يبين العمود الثاني حجم هيدروكسيد الباريوم المأخوذ في كل حالة عند معايرة 20 مل من مخلوط التفاعل .. كما يبين العمود الثالث كمية CO2 بالجرام المتحررة بواسطة التفاعل الأول. أوجدي ثوابت التفاعلين K2 , K1 بمعلومية أن عيارية هيدروكسيد الباريوم المستخدمة هي 0.1 وأن التفاعلات العكسية يمكن إهمالها؟
 حجم هيدروكسيد الباريوم المأخوذ. t ( min ) , , , حيث يبين العمود الثاني حجم هيدروكسيد الباريوم المأخوذ في كل حالة عند معايرة 20 مل من مخلوط التفاعل .. كما يبين العمود الثالث كمية CO2 بالجرام المتحررة بواسطة التفاعل الأول. أوجدي ثوابت التفاعلين K2 , K1 بمعلومية أن عيارية هيدروكسيد الباريوم المستخدمة هي 0.1 وأن التفاعلات العكسية يمكن إهمالها؟")
116
التفاعلات السلسلية تعد التفاعلات من أهم أنواع التفاعلات المعقدة، وتحدث التفاعلات السلسليه إذ استطاعت نواتج التفاعل تنشيط جزيئات أخرى من المواد المتفاعلة لتصبح مواد ناتجة وهكذا تستمر هذه الدورة ولا يتوقف التفاعل السلسلي حتى تستنفذ كل المواد المتفاعلة. وقد لوحظ توقف التفاعل السلسلي في بعض الأحيان نتيجة لمؤثرات خارجية، كان يصطدم أحد الجزيئات المنشطة في السلسلة بجدار الإناء الحاوي أو بأيه مادة غربية موجودة على هيئة شوائب .. كما أن هناك فرصة أن يتحد بجزيء آخر منشط في السلسلة مما يمنع استمرار التفاعل. ومن أمثلة التفاعلات السلسليه : التفاعل بين الكلور والهيدروجين بتأثير الضوء لتكوين حمض الهيدروكلوريك بما يعرف باتحاد الضوء-كيميائي , و يلاحظ دائماً أن التفاعل السلسلي بين الكلور والهيدروجين يتوقف عند إضافة مسحوق من الزجاج في إناء التفاعل .. كما أن الضغط يعد عاملاً مهماً لبدء أو إيقاف التفاعل السلسلي فتخفيض الضغط للتفاعل السابق يزيد من فرص وصول الذرات المتفاعلة إلى جدار الإناء وبالتالي يؤدي إلى إيقاف التفاعل أما زيادة الضغط فتقلل من عدد الذرات التي تصل إلى جدار الإناء وبالتالي تؤدي إلى استمرار التفاعل لان اصطدام الجزيئات بجدران الإناء بفقدها طاقتها الزائدة في حالة تفاعل الهيدروجين مع الكلور ويساعد ذلك على تكوين ناتج الخطوات الثلاث السابقة التي تؤدي إلى إيقاف التفاعل.

117
ولإيجاد معادلة سرعة التفاعل يتضح أن السرعة النوعية للتفاعل تعتمد من بين العوامل المختلفة الأخرى على عدد ناقلات السلسلة Chain Carriers مثل الكلور والهيدروجين في المثال السابق وعلى حجم إناء التفاعل وعلى ضغط الغازات ودرجة الحرارة، ويمكن توضيح ذلك كما يلي: - تتناسب سرعة التفاعل تناسباً طردياً مع تركيز المواد المتفاعلة : R α 1 / F1 (C) - تتناسب سرعة التفاعل تناسباً عكسيا مع سطح جدار إناء التفاعل لأن هذا يؤثر في عدد تصادمات الجزيئات الناشرة لسلسلة التفاعل مع جدار إناء التفاعل. R α 1 / F2 (S) - تتناسب سرعة التفاعل تناسباً عكسياً مع عدد مرات الاصطدام بجزيئات الغاز : R α 1 / F3 (g) والتفاعلات السلسلية ليست دائماً تتبع قوانين الكيمياء الحركية المعقدة، بل أحياناً تتبع القوانين البسيطة كما أنه غالباً ما يكون من الصعب التمييز بين تفاعل سلسلي وتفاعل عادي ويمكن أحياناً التأكد من ذلك إما بإضافة كمية معينة من الطاقة إذا كان التفاعل ضوئياً كيميائياً ثم إثبات أن عدد الجزيئات المتفاعلة أكبر بكثير من عدد الجزيئات المنشطة أو بإضافة مادة تعوق التفاعل وتوقفه تسمى بالمثبط (inhibitor) ويلاحظ أنه إذا كان جزئ من المثبط يوقف سلسلة وكانت كل سلسلة تحتوي عدد كبير من الجزيئات فأن آثاراً طفيفة من المثبط تكفي لوقف التفاعل .
 - تتناسب سرعة التفاعل تناسباً عكسياً مع عدد مرات الاصطدام بجزيئات الغاز : R α 1 / F3 (g) والتفاعلات السلسلية ليست دائماً تتبع قوانين الكيمياء الحركية المعقدة، بل أحياناً تتبع القوانين البسيطة كما أنه غالباً ما يكون من الصعب التمييز بين تفاعل سلسلي وتفاعل عادي ويمكن أحياناً التأكد من ذلك إما بإضافة كمية معينة من الطاقة إذا كان التفاعل ضوئياً كيميائياً ثم إثبات أن عدد الجزيئات المتفاعلة أكبر بكثير من عدد الجزيئات المنشطة أو بإضافة مادة تعوق التفاعل وتوقفه تسمى بالمثبط (inhibitor) ويلاحظ أنه إذا كان جزئ من المثبط يوقف سلسلة وكانت كل سلسلة تحتوي عدد كبير من الجزيئات فأن آثاراً طفيفة من المثبط تكفي لوقف التفاعل .")
118
مثال للتفاعلات السلسلية( تفاعل البروم مع غاز الهيدروجين )
مثال للتفاعلات السلسلية( تفاعل البروم مع غاز الهيدروجين ) هذا التفاعل يعد من التفاعلات السلسلية والتي تحدث نتيجة لتكون ذرات أو شقوق كحالات وسطية أثناء التفاعل، يحدث هذا التفاعل حسب الخطوات المقترحة الآتية : بدء التفاعل : Br2 K1 2Br (1) انتشار التفاعل : Br + H2 K2 HBr + H (2) H + Br2 K3 HBr + Br (3) تثبيط التفاعل : H + HBr2 K4 H2 + Br (4) كسر التفاعل السلسلي : 2Br K5 Br2 (5)
 هذا التفاعل يعد من التفاعلات السلسلية والتي تحدث نتيجة لتكون ذرات أو شقوق كحالات وسطية أثناء التفاعل، يحدث هذا التفاعل حسب الخطوات المقترحة الآتية : بدء التفاعل : Br2 K1 2Br (1) انتشار التفاعل : Br + H2 K2 HBr + H (2) H + Br2 K3 HBr + Br (3) تثبيط التفاعل : H + HBr2 K4 H2 + Br (4) كسر التفاعل السلسلي : 2Br K5 Br2 (5)")
119
d[H] /dt = K2[Br] [H2]- K3[H][Br2]-K4 [H][HBr] =0 (7)
وقد افترض في هذا التفاعل أن كلاً من ذرات البروم و ذرات الهيدروجين تتصرف كحالات وسطية، كما أنه يتم توليد عدد كبير من ذرات الهيدروجين بواسطة ذرات أو جزيئات البروم وبذلك فإن عدد الناقلات في التفاعل سوف يزداد لحساب سرعة التفاعل الكلي على ضوء ثوابت سرعة خطوات التفاعل فإنه بافتراض الحالة الثابتة تكون الحالات الوسيطة أثناء سير التفاعل ذات تركيز ثابت أي أن : d [Br] / dt = d [H] / dt = 0 وعلى ضوء هذا الفرض ومن خطوات التفاعل المقترحة يمكن حساب سرعة التفاعل الكلية كما يلي : d[Br] /dt = K1[Br2] - K2[Br][H2]+ K3[H][Br2]+K4 [H][HBr] – K5[Br]2 = 0 d[Br]/dt =K1[Br2]–{K2[Br][H2] - K3[H][Br2]-K4 [H][HBr]} – K5[Br]2 = (6) كذلك : d[H] /dt = K2[Br] [H2]- K3[H][Br2]-K4 [H][HBr] = (7) وبالتعويض من المعادلة(7 ) في المعادلة (6) ينتج أن : d[Br] /dt = K1[Br2] – 0 – K5 [Br]2 = 0
![d[H] /dt = K2[Br] [H2]- K3[H][Br2]-K4 [H][HBr] =0 (7)](http://slideplayer.ae/slide/15551337/88/images/119/%EF%81%9Cd%5BH%5D+%2Fdt+%3D+K2%5BBr%5D+%5BH2%5D-+K3%5BH%5D%5BBr2%5D-K4+%5BH%5D%5BHBr%5D+%3D0+%287%29.jpg "وقد افترض في هذا التفاعل أن كلاً من ذرات البروم و ذرات الهيدروجين تتصرف كحالات وسطية، كما أنه يتم توليد عدد كبير من ذرات الهيدروجين بواسطة ذرات أو جزيئات البروم وبذلك فإن عدد الناقلات في التفاعل سوف يزداد لحساب سرعة التفاعل الكلي على ضوء ثوابت سرعة خطوات التفاعل فإنه بافتراض الحالة الثابتة تكون الحالات الوسيطة أثناء سير التفاعل ذات تركيز ثابت أي أن : d [Br] / dt = 0 d [H] / dt = 0. وعلى ضوء هذا الفرض ومن خطوات التفاعل المقترحة يمكن حساب سرعة التفاعل الكلية كما يلي : d[Br] /dt = K1[Br2] - K2[Br][H2]+ K3[H][Br2]+K4 [H][HBr] – K5[Br]2 = 0. d[Br]/dt =K1[Br2]–{K2[Br][H2] - K3[H][Br2]-K4 [H][HBr]} – K5[Br]2 = 0 (6) كذلك : d[H] /dt = K2[Br] [H2]- K3[H][Br2]-K4 [H][HBr] =0 (7) وبالتعويض من المعادلة(7 ) في المعادلة (6) ينتج أن : d[Br] /dt = K1[Br2] – 0 – K5 [Br]2 = 0.")
120
ومنه : [Br˙]2 = K1 / K5 [Br2] [Br] = ( K1 / K5 )½ [Br2]½ (8) كذلك بالتعويض في المعادلة (7) بقيمة (Br) من المعادلة (8) يتم الحصول على : [H] = K2 ( K1 / K5 )½ [H2] [Br2]½ / K5 [Br1] + K4 [HBr] ويمكن الحصول على معدل تكوين بروميد الهيدروجين HBr من المعادلات (2)، (3)، (4) كما يلي : d[HBr] / dt = K2 [Br][H2] + K3[H][Br2] – K4[H][HBr] (9) بالتعويض عن قيمة (Br) و (H) في المعادلة (9) يتم الحصول على : d[HBr] /dt = 2K2 (K1 / K5)½ [H2][Br2]½ / 1+K4 [HBr] / K3 [Br2] (10) ويمكن كتابة هذه المعادلة على الصورة الآتية : d[HBr] /dt = K [H2][Br2]½ / 1+ [HBr] / Ḱ [Br2] (11) حيث : 2K2 ( K1 / K5 )½ تساوي K K3 / K4تساوي Ḱ
![ومنه : [Br˙]2 = K1 / K5 [Br2] [Br] = ( K1 / K5 )½ [Br2]½ (8) كذلك بالتعويض في المعادلة (7) بقيمة (Br) من المعادلة (8) يتم الحصول على : [H] = K2 ( K1 / K5 )½ [H2] [Br2]½ / K5 [Br1] + K4 [HBr] ويمكن الحصول على معدل تكوين بروميد الهيدروجين HBr من المعادلات (2)، (3)، (4) كما يلي : d[HBr] / dt = K2 [Br][H2] + K3[H][Br2] – K4[H][HBr] (9) بالتعويض عن قيمة (Br) و (H) في المعادلة (9) يتم الحصول على : d[HBr] /dt = 2K2 (K1 / K5)½ [H2][Br2]½ / 1+K4 [HBr] / K3 [Br2] (10) ويمكن كتابة هذه المعادلة على الصورة الآتية : d[HBr] /dt = K [H2][Br2]½ / 1+ [HBr] / Ḱ [Br2] (11) حيث : 2K2 ( K1 / K5 )½ تساوي K K3 / K4تساوي Ḱ](http://slideplayer.ae/slide/15551337/88/images/120/%D9%88%D9%85%D9%86%D9%87+%3A+%5BBr%CB%99%5D2+%3D+K1+%2F+K5+%5BBr2%5D+%EF%81%9C%5BBr%5D+%3D+%28+K1+%2F+K5+%29%C2%BD+%5BBr2%5D%C2%BD+%288%29+%D9%83%D8%B0%D9%84%D9%83+%D8%A8%D8%A7%D9%84%D8%AA%D8%B9%D9%88%D9%8A%D8%B6+%D9%81%D9%8A+%D8%A7%D9%84%D9%85%D8%B9%D8%A7%D8%AF%D9%84%D8%A9+%287%29+%D8%A8%D9%82%D9%8A%D9%85%D8%A9+%28Br%29+%D9%85%D9%86+%D8%A7%D9%84%D9%85%D8%B9%D8%A7%D8%AF%D9%84%D8%A9+%288%29+%D9%8A%D8%AA%D9%85+%D8%A7%D9%84%D8%AD%D8%B5%D9%88%D9%84+%D8%B9%D9%84%D9%89+%3A+%5BH%5D+%3D+K2+%28+K1+%2F+K5+%29%C2%BD+%5BH2%5D+%5BBr2%5D%C2%BD+%2F+K5+%5BBr1%5D+%2B+K4+%5BHBr%5D+%D9%88%D9%8A%D9%85%D9%83%D9%86+%D8%A7%D9%84%D8%AD%D8%B5%D9%88%D9%84+%D8%B9%D9%84%D9%89+%D9%85%D8%B9%D8%AF%D9%84+%D8%AA%D9%83%D9%88%D9%8A%D9%86+%D8%A8%D8%B1%D9%88%D9%85%D9%8A%D8%AF+%D8%A7%D9%84%D9%87%D9%8A%D8%AF%D8%B1%D9%88%D8%AC%D9%8A%D9%86+HBr+%D9%85%D9%86+%D8%A7%D9%84%D9%85%D8%B9%D8%A7%D8%AF%D9%84%D8%A7%D8%AA+%282%29%D8%8C+%283%29%D8%8C+%284%29+%D9%83%D9%85%D8%A7+%D9%8A%D9%84%D9%8A+%3A+d%5BHBr%5D+%2F+dt+%3D+K2+%5BBr%5D%5BH2%5D+%2B+K3%5BH%5D%5BBr2%5D+%E2%80%93+K4%5BH%5D%5BHBr%5D+%289%29+%D8%A8%D8%A7%D9%84%D8%AA%D8%B9%D9%88%D9%8A%D8%B6+%D8%B9%D9%86+%D9%82%D9%8A%D9%85%D8%A9+%28Br%29+%D9%88+%28H%29+%D9%81%D9%8A+%D8%A7%D9%84%D9%85%D8%B9%D8%A7%D8%AF%D9%84%D8%A9+%289%29+%D9%8A%D8%AA%D9%85+%D8%A7%D9%84%D8%AD%D8%B5%D9%88%D9%84+%D8%B9%D9%84%D9%89+%3A+d%5BHBr%5D+%2Fdt+%3D+2K2+%28K1+%2F+K5%29%C2%BD+%5BH2%5D%5BBr2%5D%C2%BD+%2F+1%2BK4+%5BHBr%5D+%2F+K3+%5BBr2%5D+%2810%29+%D9%88%D9%8A%D9%85%D9%83%D9%86+%D9%83%D8%AA%D8%A7%D8%A8%D8%A9+%D9%87%D8%B0%D9%87+%D8%A7%D9%84%D9%85%D8%B9%D8%A7%D8%AF%D9%84%D8%A9+%D8%B9%D9%84%D9%89+%D8%A7%D9%84%D8%B5%D9%88%D8%B1%D8%A9+%D8%A7%D9%84%D8%A2.jpg "ومنه : [Br˙]2 = K1 / K5 [Br2] [Br] = ( K1 / K5 )½ [Br2]½ (8) كذلك بالتعويض في المعادلة (7) بقيمة (Br) من المعادلة (8) يتم الحصول على : [H] = K2 ( K1 / K5 )½ [H2] [Br2]½ / K5 [Br1] + K4 [HBr] ويمكن الحصول على معدل تكوين بروميد الهيدروجين HBr من المعادلات (2)، (3)، (4) كما يلي : d[HBr] / dt = K2 [Br][H2] + K3[H][Br2] – K4[H][HBr] (9) بالتعويض عن قيمة (Br) و (H) في المعادلة (9) يتم الحصول على : d[HBr] /dt = 2K2 (K1 / K5)½ [H2][Br2]½ / 1+K4 [HBr] / K3 [Br2] (10) ويمكن كتابة هذه المعادلة على الصورة الآتية : d[HBr] /dt = K [H2][Br2]½ / 1+ [HBr] / Ḱ [Br2] (11) حيث : 2K2 ( K1 / K5 )½ تساوي K K3 / K4تساوي Ḱ")
121
حركة التفاعلات الكيميائية من الوجهة النظرية (نظريات سرعات التفاعل)
حركة التفاعلات الكيميائية من الوجهة النظرية (نظريات سرعات التفاعل) تهدف دراسة سرعة التفاعلات الكيميائية إلى التعرف على المعلومات الأساسية عن صفات الجزيئات المتفاعلة. ويهتم هذا الجزء بدراسة نظريتين تهتمان بحساب طاقة التنشيط التي ترتبط ارتباطاً واضحاً بسرعة التفاعلات الكيميائية حسب معادلة أرهينـوس التي يمكن أن تكتب على أي من الصورتين: K = Ae –ΔH/RT K = Ae-ΔE/RT وهاتان النظريتان هما: - نظرية التصادم. - نظرية سرعة التفاعلات المطلقة. وقيم ΔH المحسوبة من هاتين النظريتين هي في حدود التقريب المقبول لأن الحساب الدقيق للطاقة التنشيطية بالطريقة النظرية يكون مشكلة في غاية التعقيد وأمكن تطبيقها فقط لعدد محدود من التفاعلات السهلة.
 تهدف دراسة سرعة التفاعلات الكيميائية إلى التعرف على المعلومات الأساسية عن صفات الجزيئات المتفاعلة. ويهتم هذا الجزء بدراسة نظريتين تهتمان بحساب طاقة التنشيط التي ترتبط ارتباطاً واضحاً بسرعة التفاعلات الكيميائية حسب معادلة أرهينـوس التي يمكن أن تكتب على أي من الصورتين: K = Ae –ΔH/RT K = Ae-ΔE/RT وهاتان النظريتان هما: - نظرية التصادم. - نظرية سرعة التفاعلات المطلقة. وقيم ΔH المحسوبة من هاتين النظريتين هي في حدود التقريب المقبول لأن الحساب الدقيق للطاقة التنشيطية بالطريقة النظرية يكون مشكلة في غاية التعقيد وأمكن تطبيقها فقط لعدد محدود من التفاعلات السهلة.")
122
Activation Energy الطاقة التنشيطية
لكي تتمكن الجزيئات من التفاعل فإنها تحتاج إلى مقدار من الطاقة يختلف باختلاف الجزيئات نفسها، واختلاف التفاعلات التي تشترك فيها، هذه الطاقة تسمى بالطاقة التنشيطية. وحيث أن الكيمياء الحركية تهتم بميكانيكية التفاعل والخطوات المتوسطة، أي بخطوات التفاعل كلها التي تؤدي إلى النواتج النهائية، لذلك يجب عند تطبيق حسابات الديناميكا الحرارية أن تطبق على التفاعل بكامل خطواته وأن لا تـقـتصر على الحالات الابتدائية والنهائية، كما هي الحال في دراسة الاتزان الكيميائي والديناميكا الحرارية. ولما كانت سرعة التفاعل تحددها الخطوة البطيئة والتي تعرف بالخطوة المحددة لسرعة التفاعل (Rate determining step) فإن الكيمياء الحركية تهتم بصفة خاصة بحساب طاقة التنشيط لهذه الخطوة. وتعد كمية الطاقة اللازمة للحالة المنشطة العامل الأساسي في تعيين سرعة التفاعل وليس للحرارة المنطلقة في تفاعل ما أية أهمية في حساب سرعات التفاعل، ولذلك فأنه كلما كانت الطاقة اللازمة للتنشيط كبيرة كان التفاعل بطيئاً، بينما في التفاعلات السريعة تكون طاقة التنشيط اللازمة لحدوث التفاعل صغيرة جداً.
 فإن الكيمياء الحركية تهتم بصفة خاصة بحساب طاقة التنشيط لهذه الخطوة. وتعد كمية الطاقة اللازمة للحالة المنشطة العامل الأساسي في تعيين سرعة التفاعل وليس للحرارة المنطلقة في تفاعل ما أية أهمية في حساب سرعات التفاعل، ولذلك فأنه كلما كانت الطاقة اللازمة للتنشيط كبيرة كان التفاعل بطيئاً، بينما في التفاعلات السريعة تكون طاقة التنشيط اللازمة لحدوث التفاعل صغيرة جداً.")
123
ولا يمكن قياس حرارة التنشيط مباشرة باســتخدام المسعر لأن الجزيئات المنشطة ليس لها إلا وجود قصير جداً، ولكنها تعين بطريقة غير مباشرة، وذلك برسم log K مقابل 1/T وضرب الميل بمقدار (2.303 R) أي بالطريقة نفسها التي تعين بها حرارة التفاعل برسم log K مقابل 1/T حيث K هنا هو ثابت الاتزان ويساوي Kf /Kr (النسبة بين ثابتي التفاعل في الاتجاه الطردي والعكسي) ومن معادلة فانت هوف: d (ln K)/dT = ΔHº/RT2 حيث ΔHº هي طاقة التنشيط للمواد المتفاعلة في الحالة القياسية، بفصل المتغيرات وبتكامل هذه المعادلة وحساب ثابت التكامل يتم الحصول على: ln K= ΔHº/RT + ln Kα حيث α ln K هي ثابت التكامل وتساوي (Kf)α/ (Kr)α (النسبة بين ثابتي التفاعل الطردي والعكسي في الحالة القياسية عند الصفر المطلق). ولكن : ΔHº = HºP - HºR حيث HºP هي الحرارة الكلية للمواد الناتجة. HºR هي الحرارة الكلية للمواد المتفاعلة. بالتعويض عن كل من Kα ,K, ΔHº في المعادلة (55) بما يساويهم ينتج أن: ] ln Kf/Kr = -(HºP-HºR/RT) + ln[(Kf) α/(Kr) α Or ln Kf/(Kf) α - HºR/RT = ln Kr/(Kr) α - HºP/RT (56)
 بما يساويهم ينتج أن: ] ln Kf/Kr = -(HºP-HºR/RT) + ln[(Kf) α/(Kr) α Or ln Kf/(Kf) α - HºR/RT = ln Kr/(Kr) α - HºP/RT (56) .")
124
يلاحظ في المعادلة (56) أن الطرف الأيسر يعتمد فقط على صفات المواد المتفاعلة، بينما الطرف الأيمن كما يظهر يعتمد على صفات المواد الناتجة، وعلى هذا فكلا الطرفين يجب أن يساوي مقداراً ثابتاً يمكن التعبير عنه بالمقدار –ΔH*/RT وعلى هذا فالمعادلة (56) يمكن كتابتها على النحو التالي: ln Kf/(Hf) α = -H*-HºR/RT (57) ln Kr/(Kr) α = -H*-HºP/RT (58) ومن هذا يمكن الاستنتاج أن: Kf = (Kf)α e - (H*- HºR /RT) (59) Kr = (Kr)α e - (H*- HºP /RT) (60) أي انه يمكن برهان معادلة اهينوس لثابت التفاعل بالنسبة إلى أي تفاعل كيميائي أولي في أي إتجاه، المقدار (H*-HºR) يمثل مقدار الحرارة الكلية في معادلة أرهينوس وتعطي الرمز H*f وقد وجد عملياً أن H*f هي كمية موجبة، ومعنى هذا المقدار (H*-HºR) موجب أيضاً، أي أن HºR<H* وبالطريقة نفسها فأن HºP<H*.
 يمكن كتابتها على النحو التالي: ln Kf/(Hf) α = -H*-HºR/RT (57) ln Kr/(Kr) α = -H*-HºP/RT (58) ومن هذا يمكن الاستنتاج أن: Kf = (Kf)α e - (H*- HºR /RT) (59) Kr = (Kr)α e - (H*- HºP /RT) (60) أي انه يمكن برهان معادلة اهينوس لثابت التفاعل بالنسبة إلى أي تفاعل كيميائي أولي في أي إتجاه، المقدار (H*-HºR) يمثل مقدار الحرارة الكلية في معادلة أرهينوس وتعطي الرمز H*f وقد وجد عملياً أن H*f هي كمية موجبة، ومعنى هذا المقدار (H*-HºR) موجب أيضاً، أي أن HºR<H* وبالطريقة نفسها فأن HºP<H*.")
125
التغيير في مقدار الحرارة الكلية (Enthaply) خلال الخطوة الأولى لتحويل المواد المتفاعلة إلى مواد ناتجة يمكن توضيحه في الشكل الآتي: يبين الشكل(4): تغير الحرارة الكلية خلال حدوث التفاعل. ووفقاً لوجهة النظر بالنسبة إلى هذه الحالة يمكن اعـتبار حاجز الطاقة Barrier Energy هو الذي يفصل حالة المادة المتفاعلة عن حالة المادة الناتجة، وعند تصادم المواد المتفاعلة يجب أن تكتسب مقداراً من الحرارة الكلية يمكنها من التغلب على حاجز الطاقة حتى يمكن أن تتحول إلى مواد ناتجة وإلا فلو كانت جميع الجزيئات متساوية في فعاليتها لكان من الصعب تفسير وجود التفاعلات البطيئة. ولما كان عدد التصادمات في الثانية كبيراً جداً فإن جميع التصادمات لا تؤدي إلى تفاعلات كيميائية وإنما هذا يعتمد على مقدار الطاقة المكتسبة وحتى يسير التفاعل في الاتجاه الأمامي فأن طاقة التنشيط اللازمة يمثلها إرتفاع حاجز الطاقة وفي هذه الحالة هو (H*-HºR). المركب النشيط

126
ويمكن الإشارة إلى طاقة التنشيط بالرمز E. f وهناك فرق بين H. f ,E
ويمكن الإشارة إلى طاقة التنشيط بالرمز E*f وهناك فرق بين H*f ,E*f والعلاقة بينهما تعتمد على نوع التفاعل. الجزيئات المتفاعلة التي تكتسب عند تصادمها مقدارها من الطاقة أقل من المطلوب للتغلب على حاجز الطاقة تبقى على حالتها جزيئات متفاعلة، أما تلك التي تكتسب مقداراً من الطاقة يمكنها من التغلب على حاجز الطاقة فيتحول معظمها إلى مواد ناتجة. أما إذا كان التفاعل عكسياً فإن حاجز الطاقة بالنسبة للمواد الناتجة هو H*-HºP وهذا المقدار هو طاقة التنشيط للتفاعل في الاتجاه المعاكس ويمكن الإشارة إليه بالرمز E*r العلاقة بين طاقة التنشيط للحالتين يمكن الحصول عليها كما يلي: H*-HºP = H*- HºR+ HºR -HºP = H*-HºR-∆Hº H*-HºP = H*- HºR - ∆Hº E*r = E*f - ∆Hº (61) وهذه المعادلة تمثل العلاقة العامة لطاقات التنشيط والتغير في الطاقة لأي تفاعل، فإذا عرفت قيمة طاقة التنشيط للتفاعل في الاتجاه الأمامي وعرفت قيمة ∆Hº امكن حساب طاقة التنشيط للتفاعل في الاتجاه العكسي مباشرة.
 وهذه المعادلة تمثل العلاقة العامة لطاقات التنشيط والتغير في الطاقة لأي تفاعل، فإذا عرفت قيمة طاقة التنشيط للتفاعل في الاتجاه الأمامي وعرفت قيمة ∆Hº امكن حساب طاقة التنشيط للتفاعل في الاتجاه العكسي مباشرة.")
127
نظريات سرعة التفاعل هناك نظريتان أساسيتان لتفسير سرعات التفاعلات الكيميائية وهما: نظرية التصادم. نظرية سرعة التفاعلات الطلقة (نظرية الحالة الانتقائية). أولاً: نظرية التصادم: تعتمد هذه النظرية أساساً على النظرية الحركية للغازات، وتطبق بدرجة فعالة على التفاعلات الثنائية البسيطة وإذا أضيفت إليها بعض الافتراضات يمكن تطبيقها على التفاعلات من الدرجة الأولى. فإذا فرض أن جزيئين اتحدا كيميائياً فلابد أن تكون الخطوة الأولى لهذا الاتحاد أن يحدث تقابل أو تصادم بين هذين الجزيئين وباستعمال رأي أرهينيوس في طاقة التنشيط وهو ليس كل التصادمات تؤدي إلى تفاعل ولكن فقط التصادمات التي تحصل على اكبر قدر من طاقة التنشيط هي التي يمكنها أن تتم التفاعل ويمكن التعبير عن نظرية التصادم في جملة هي: (سرعة التفاعل تساوي عدد التصادمات النشيطة في وحدة الزمن) ولشرح هذه النظرية يفترض التفاعل الثنائي البسيط الآتي بين الجزأين :
. أولاً: نظرية التصادم: تعتمد هذه النظرية أساساً على النظرية الحركية للغازات، وتطبق بدرجة فعالة على التفاعلات الثنائية البسيطة وإذا أضيفت إليها بعض الافتراضات يمكن تطبيقها على التفاعلات من الدرجة الأولى. فإذا فرض أن جزيئين اتحدا كيميائياً فلابد أن تكون الخطوة الأولى لهذا الاتحاد أن يحدث تقابل أو تصادم بين هذين الجزيئين وباستعمال رأي أرهينيوس في طاقة التنشيط وهو ليس كل التصادمات تؤدي إلى تفاعل ولكن فقط التصادمات التي تحصل على اكبر قدر من طاقة التنشيط هي التي يمكنها أن تتم التفاعل ويمكن التعبير عن نظرية التصادم في جملة هي: (سرعة التفاعل تساوي عدد التصادمات النشيطة في وحدة الزمن) ولشرح هذه النظرية يفترض التفاعل الثنائي البسيط الآتي بين الجزأين :")
128
A + B AB -dnA/dt = -dnB /dt = ZAB e-E/RT (62)
حيث nA , nB عدد جزيئات المواد المتفاعلة B , A في وحدة الحجم. ZAB = عدد الصدمات بين B , A في وحدة الحجم في وحدة الزمن - dn/dt = النقص في عدد الجزيئات B , A في وحدة الزمن (سرعة التفاعل). حيث dn/dt , ZAB ليست قيما ثابتة لأن عدد الجزيئات المتفاعلة تنقص كلما تقدم التفاعل ومن تعريف ثابت السرعة لتفاعلات الدرجة الثانية يتم الحصول على: -dn/dt = K nA nB (63) حيث nA , nB عدد الجزيئات في وحدة الحجم للمادتين B , A وقد عــــــبر عن ثابـــت التفاعل K بالوحدات المناسبة. من المعادلتين (62) و (63) ينتج أن: ZABe-E/RT = K nA nB K= ZAB/nA nB e-E/RT Or K= Z- e-E/RT
. حيث dn/dt , ZAB ليست قيما ثابتة لأن عدد الجزيئات المتفاعلة تنقص كلما تقدم التفاعل ومن تعريف ثابت السرعة لتفاعلات الدرجة الثانية يتم الحصول على: -dn/dt = K nA nB (63) حيث nA , nB عدد الجزيئات في وحدة الحجم للمادتين B , A وقد عــــــبر عن ثابـــت التفاعل K بالوحدات المناسبة. من المعادلتين (62) و (63) ينتج أن: ZABe-E/RT = K nA nB. K= ZAB/nA nB e-E/RT. Or. K= Z- e-E/RT.")
129
حيث Z- تسمى ثابت التصادم أو عدد التصادم
حيث Z- تسمى ثابت التصادم أو عدد التصادم. Z- = ZAB/ nA nB (64) المعادلة (64) تعطي المعنى الرياضي لنظرية التصادم لسرعات التفاعل ويلاحظ أن هذه المعادلة تشبه معادلة أرهينوس. K=A e ∆H/RT وذلك باستبدال القيمة Z- بالقيمة A حيث كلاً من H , E تمثل طاقة التنشيط أي أن نظرية التصادم تتنبأ بعامل التردد A. ولتعيين قيمة الثابت Z- وبالتالي عامل أرهينوس A يتم التعويض أولاً عن قيمة المقدار ZAB وهو عدد الصدمات في وحدة الحجم في وحدة الزمن وذلك من النظرية الحركية للغازات أي: ZAB = (σA+ σB/2)2 √8π(mA+ mB) kT/mA mB nA nB حيث B , σAσ هي أقطار الجزيئات B , A mB , mA هي كتلة الجزيئات B , A nB , nA هي عدد الجزيئات B , A في السنتيمتر المكعب. K هي ثابت بولتزمان R/N= ، حيث N عدد أفـوجادرو.
 المعادلة (64) تعطي المعنى الرياضي لنظرية التصادم لسرعات التفاعل ويلاحظ أن هذه المعادلة تشبه معادلة أرهينوس. K=A e ∆H/RT وذلك باستبدال القيمة Z- بالقيمة A حيث كلاً من H , E تمثل طاقة التنشيط أي أن نظرية التصادم تتنبأ بعامل التردد A. ولتعيين قيمة الثابت Z- وبالتالي عامل أرهينوس A يتم التعويض أولاً عن قيمة المقدار ZAB وهو عدد الصدمات في وحدة الحجم في وحدة الزمن وذلك من النظرية الحركية للغازات أي: ZAB = (σA+ σB/2)2 √8π(mA+ mB) kT/mA mB nA nB حيث B , σAσ هي أقطار الجزيئات B , A mB , mA هي كتلة الجزيئات B , A nB , nA هي عدد الجزيئات B , A في السنتيمتر المكعب. K هي ثابت بولتزمان R/N= ، حيث N عدد أفـوجادرو.")
130
بالتعويض في المعادلة (64) نجد أن: Z- =(σA+ σB/2)2 √8π(mA+mB) kT/ mA mB = A (65) أي أن عامل التردد A (قيمة ثابت التصادم) تعتمد على الجذر التربيعي لدرجة الحرارة وهو تأثير طفيف إذ المفروض أن يكون عامل التردد A مستقلاً عن درجة الحرارة، قيمة الجذر في المعادلة (65) تعبر عن سرعة الجزيئات في درجة الحرارة العادية وتقدر بحوالي 4x410 سم/ثانية وقيمة σ في حدود 10-8 وبهذا فأن: A = (10-8)2 (4x104) = 4x1012 cm3/atom/sec وحيث أن وحدة التركيز هي وزن جزئ في اللتر فإنه: A = 4x10-12 x N/1000 = 4x10-12 x 6x1023/1000 = 2.4x109 L/mol/sec رتبة كمية عامل التردد للتفاعلات الثنائية هي بين 910 ، 1010، إذا أخذت وحدة التركيز جزئياً في اللتر، وكانت درجة الحرارة في حدود ◦300 درجة مطلقة وقد وجد أن عامل التردد للتفاعلات التي تشمل جزيئات خفيفة مثل الهيدروجين عادة تكون اكبر (في حدود 1110) .
 نجد أن: Z- =(σA+ σB/2)2 √8π(mA+mB) kT/ mA mB = A (65) أي أن عامل التردد A (قيمة ثابت التصادم) تعتمد على الجذر التربيعي لدرجة الحرارة وهو تأثير طفيف إذ المفروض أن يكون عامل التردد A مستقلاً عن درجة الحرارة، قيمة الجذر في المعادلة (65) تعبر عن سرعة الجزيئات في درجة الحرارة العادية وتقدر بحوالي 4x410 سم/ثانية وقيمة σ في حدود 10-8 وبهذا فأن: A = (10-8)2 (4x104) = 4x1012 cm3/atom/sec وحيث أن وحدة التركيز هي وزن جزئ في اللتر فإنه: A = 4x10-12 x N/1000 = 4x10-12 x 6x1023/1000 = 2.4x109 L/mol/sec رتبة كمية عامل التردد للتفاعلات الثنائية هي بين 910 ، 1010، إذا أخذت وحدة التركيز جزئياً في اللتر، وكانت درجة الحرارة في حدود ◦300 درجة مطلقة وقد وجد أن عامل التردد للتفاعلات التي تشمل جزيئات خفيفة مثل الهيدروجين عادة تكون اكبر (في حدود 1110) .")
131
نظرية التصادم تستطيع أن تتنبأ بنجاح بقيمة ثابت سرعة التفاعل للتفاعلات التي تشمل نسبياً جزيئات بسيطة إذا عرف مقدار طاقة التنشيط ولكن الصعوبة تقابل التفاعلات التي تشمل الجزيئات الـمعقدة لأن ســرعة التــفاعل تكون أقل مما تتنبأ به نظـــرية التـــصادم، وفي أكثر الحالات بمقدار 10 درجات أو أكثر وللتغلب على هذا الحيود يضاف عامل جديد (P) يسمى بمعامل الاحتمال فتصبح المعادلة (64) والتي تعطي المعنى الرياضي لنظرية التصادم كالتالي: K=PZ e-E*/RT (66) والسبب في وجود العامل (P) أنه حتى التصــادمات التي تحصل على طاقة التنشيط الكافية يمكن أن لا تؤدي إلى التفاعل وفي الأصل كان الافتراض الأول لم يوضح أن الجزيئات يجب أن تصطدم على هيئة خاصة ولهذا أدخل عامل الاحتمال، هذا الافتراض قابل للتطبيق ولاسيما أن انخفاض سرعة التفاعل تلاحظ عادة مع الجزيئات المعقدة وغالباً أن الجزيئين المعقدين فرصتهما في التصادم أقل من الجزئيين البسيطين.
 يسمى بمعامل الاحتمال فتصبح المعادلة (64) والتي تعطي المعنى الرياضي لنظرية التصادم كالتالي: K=PZ e-E*/RT (66) والسبب في وجود العامل (P) أنه حتى التصــادمات التي تحصل على طاقة التنشيط الكافية يمكن أن لا تؤدي إلى التفاعل وفي الأصل كان الافتراض الأول لم يوضح أن الجزيئات يجب أن تصطدم على هيئة خاصة ولهذا أدخل عامل الاحتمال، هذا الافتراض قابل للتطبيق ولاسيما أن انخفاض سرعة التفاعل تلاحظ عادة مع الجزيئات المعقدة وغالباً أن الجزيئين المعقدين فرصتهما في التصادم أقل من الجزئيين البسيطين.")
132
تطبيق نظرية التصادم: التفاعل بين ثلاثة جزيئات: لدراسة التفاعل بين ثلاثة جزيئات على ضوء نظرية التصادم ولاسيما في التفاعلات المعروفة يدرس التفاعل بين أول أكسيد النتروجين والأكسجين لتكوين ثاني أكسيد النتروجين، معادلة التفاعل هي: 2NO + O2 2NO2 ومعادلة سرعة التفاعل هي: -dC[O2 ]/dt = K[NO]2[O2] حيث K هو ثابت سرعة التفاعل [O2] [NO] هما تركيز أكسيد النتروجين والأكسجين على التوالي. هذا التفاعل كما يبدو من معادلته أنه تفاعل أولي وأنه يتطلب تصادم جزيئين من أول أكسيد النتروجين مع جزئ من الأكسجين ولكن هذا التفاعل يتمتع بظاهرة تلف النظر وهي أن سرعة تفاعله تنقص بالزيادة في درجة حرارة التفاعل، وهناك قليل من التفاعلات التي تشارك في هذه الظاهرة.
![تطبيق نظرية التصادم: التفاعل بين ثلاثة جزيئات: لدراسة التفاعل بين ثلاثة جزيئات على ضوء نظرية التصادم ولاسيما في التفاعلات المعروفة يدرس التفاعل بين أول أكسيد النتروجين والأكسجين لتكوين ثاني أكسيد النتروجين، معادلة التفاعل هي: 2NO + O2 2NO2 ومعادلة سرعة التفاعل هي: -dC[O2 ]/dt = K[NO]2[O2] حيث K هو ثابت سرعة التفاعل [O2] [NO] هما تركيز أكسيد النتروجين والأكسجين على التوالي.](http://slideplayer.ae/slide/15551337/88/images/132/%D8%AA%D8%B7%D8%A8%D9%8A%D9%82+%D9%86%D8%B8%D8%B1%D9%8A%D8%A9+%D8%A7%D9%84%D8%AA%D8%B5%D8%A7%D8%AF%D9%85%3A+%D8%A7%D9%84%D8%AA%D9%81%D8%A7%D8%B9%D9%84+%D8%A8%D9%8A%D9%86+%D8%AB%D9%84%D8%A7%D8%AB%D8%A9+%D8%AC%D8%B2%D9%8A%D8%A6%D8%A7%D8%AA%3A+%D9%84%D8%AF%D8%B1%D8%A7%D8%B3%D8%A9+%D8%A7%D9%84%D8%AA%D9%81%D8%A7%D8%B9%D9%84+%D8%A8%D9%8A%D9%86+%D8%AB%D9%84%D8%A7%D8%AB%D8%A9+%D8%AC%D8%B2%D9%8A%D8%A6%D8%A7%D8%AA+%D8%B9%D9%84%D9%89+%D8%B6%D9%88%D8%A1+%D9%86%D8%B8%D8%B1%D9%8A%D8%A9+%D8%A7%D9%84%D8%AA%D8%B5%D8%A7%D8%AF%D9%85+%D9%88%D9%84%D8%A7%D8%B3%D9%8A%D9%85%D8%A7+%D9%81%D9%8A+%D8%A7%D9%84%D8%AA%D9%81%D8%A7%D8%B9%D9%84%D8%A7%D8%AA+%D8%A7%D9%84%D9%85%D8%B9%D8%B1%D9%88%D9%81%D8%A9+%D9%8A%D8%AF%D8%B1%D8%B3+%D8%A7%D9%84%D8%AA%D9%81%D8%A7%D8%B9%D9%84+%D8%A8%D9%8A%D9%86+%D8%A3%D9%88%D9%84+%D8%A3%D9%83%D8%B3%D9%8A%D8%AF+%D8%A7%D9%84%D9%86%D8%AA%D8%B1%D9%88%D8%AC%D9%8A%D9%86+%D9%88%D8%A7%D9%84%D8%A3%D9%83%D8%B3%D8%AC%D9%8A%D9%86+%D9%84%D8%AA%D9%83%D9%88%D9%8A%D9%86+%D8%AB%D8%A7%D9%86%D9%8A+%D8%A3%D9%83%D8%B3%D9%8A%D8%AF+%D8%A7%D9%84%D9%86%D8%AA%D8%B1%D9%88%D8%AC%D9%8A%D9%86%D8%8C+%D9%85%D8%B9%D8%A7%D8%AF%D9%84%D8%A9+%D8%A7%D9%84%D8%AA%D9%81%D8%A7%D8%B9%D9%84+%D9%87%D9%8A%3A+2NO+%2B+O2+2NO2+%D9%88%D9%85%D8%B9%D8%A7%D8%AF%D9%84%D8%A9+%D8%B3%D8%B1%D8%B9%D8%A9+%D8%A7%D9%84%D8%AA%D9%81%D8%A7%D8%B9%D9%84+%D9%87%D9%8A%3A+-dC%5BO2+%5D%2Fdt+%3D+K%5BNO%5D2%5BO2%5D+%D8%AD%D9%8A%D8%AB+K+%D9%87%D9%88+%D8%AB%D8%A7%D8%A8%D8%AA+%D8%B3%D8%B1%D8%B9%D8%A9+%D8%A7%D9%84%D8%AA%D9%81%D8%A7%D8%B9%D9%84+%5BO2%5D+%5BNO%5D+%D9%87%D9%85%D8%A7+%D8%AA%D8%B1%D9%83%D9%8A%D8%B2+%D8%A3%D9%83%D8%B3%D9%8A%D8%AF+%D8%A7%D9%84%D9%86%D8%AA%D8%B1%D9%88%D8%AC%D9%8A%D9%86+%D9.jpg "هذا التفاعل كما يبدو من معادلته أنه تفاعل أولي وأنه يتطلب تصادم جزيئين من أول أكسيد النتروجين مع جزئ من الأكسجين ولكن هذا التفاعل يتمتع بظاهرة تلف النظر وهي أن سرعة تفاعله تنقص بالزيادة في درجة حرارة التفاعل، وهناك قليل من التفاعلات التي تشارك في هذه الظاهرة.")
133
الصعوبة في معالجة هذا التفاعل على ضوء نظرية التصادم تظهر بمجرد المحاولة لتعريف التصادم الثلاثي، فإذا كانت الجزيئات على هيئة كرات صلبة فإن وقت التلامس لها يكون صفراً، وعلى هذا يكون احتمال اصطدامها بـجزئ ثالث صفراً أيضاً. من أجل أن يتم تصادم جزئ ثالث مع جزيئين متصادمين في اللحظة نفسها فإن هذا يتطلب أن يستمر تلامسها بعد التصادم برهة من الزمن مهما كانت متناهية في الصغر هذه البرهة من الزمن المتناهية في الصغر افتراضاً يمكن تحديدها بالزمن الذي تظل فيه المسافة بين الجزيئين أقل من قطر الجزيئيء وبهذا يكونان في حالة تصادم وتلامس وبالحساب التقريبي لهذا الزمن وجد أن هناك احتمال وجود تصادم ثلاثي في كل عشرة الاف تصادم ثنائي عادي وهذا يفترض أن تكون التفاعلات التي تتطلب ثلاثة تصادمات تفاعلات بطيئة. وبحساب ثابت سرعة التفاعل على أساس التصادمات الثلاثية وجد أن قيمته أكبر بكثير من القيمة المتحصل عليها بالتجربة مما يعني أن عامل الاحتمال (P) يكون صغيراً.
 يكون صغيراً.")
134
وقد تم افتراض ميكانيكية بديلة لهذا النوع من التفاعلات كما يلي: يحدث التوازن التالي أولاً: NO + X2 NOX2 حيث X2 تمثل الأكسجين أو أي غاز آخر ، معادلة سرعة هذه الخطوة هي: d(NOX2)/dt = K1[NO] [X2] حيث K1 هي ثابت السرعة لهذه الخطوة يتبع ذلك التفاعل البطئ الآتي: NOX2 + NO 2NOX ومعادلة السرعة النهائية هي: R=K2[NO][NOX2] =K2K1[NO]2[X2] = K[NO]2 [X2] وهذه الميكانيكية تأخذ ي الاعتبار معادلة سرعة التفاعل ومن الواضح أن التوازن الذي تتبعه الخطوة البطيئة يمكن ان يكون كالتالي: 2NO N2O2 N2O2 + X2 2NOX
![وقد تم افتراض ميكانيكية بديلة لهذا النوع من التفاعلات كما يلي: يحدث التوازن التالي أولاً: NO + X2 NOX2 حيث X2 تمثل الأكسجين أو أي غاز آخر ، معادلة سرعة هذه الخطوة هي: d(NOX2)/dt = K1[NO] [X2] حيث K1 هي ثابت السرعة لهذه الخطوة يتبع ذلك التفاعل البطئ الآتي: NOX2 + NO 2NOX ومعادلة السرعة النهائية هي: R=K2[NO][NOX2] =K2K1[NO]2[X2] = K[NO]2 [X2] وهذه الميكانيكية تأخذ ي الاعتبار معادلة سرعة التفاعل ومن الواضح أن التوازن الذي تتبعه الخطوة البطيئة يمكن ان يكون كالتالي: 2NO N2O2 N2O2 + X2 2NOX](http://slideplayer.ae/slide/15551337/88/images/134/%D9%88%D9%82%D8%AF+%D8%AA%D9%85+%D8%A7%D9%81%D8%AA%D8%B1%D8%A7%D8%B6+%D9%85%D9%8A%D9%83%D8%A7%D9%86%D9%8A%D9%83%D9%8A%D8%A9+%D8%A8%D8%AF%D9%8A%D9%84%D8%A9+%D9%84%D9%87%D8%B0%D8%A7+%D8%A7%D9%84%D9%86%D9%88%D8%B9+%D9%85%D9%86+%D8%A7%D9%84%D8%AA%D9%81%D8%A7%D8%B9%D9%84%D8%A7%D8%AA+%D9%83%D9%85%D8%A7+%D9%8A%D9%84%D9%8A%3A+%D9%8A%D8%AD%D8%AF%D8%AB+%D8%A7%D9%84%D8%AA%D9%88%D8%A7%D8%B2%D9%86+%D8%A7%D9%84%D8%AA%D8%A7%D9%84%D9%8A+%D8%A3%D9%88%D9%84%D8%A7%D9%8B%3A+NO+%2B+X2+NOX2+%D8%AD%D9%8A%D8%AB+X2+%D8%AA%D9%85%D8%AB%D9%84+%D8%A7%D9%84%D8%A3%D9%83%D8%B3%D8%AC%D9%8A%D9%86+%D8%A3%D9%88+%D8%A3%D9%8A+%D8%BA%D8%A7%D8%B2+%D8%A2%D8%AE%D8%B1+%D8%8C+%D9%85%D8%B9%D8%A7%D8%AF%D9%84%D8%A9+%D8%B3%D8%B1%D8%B9%D8%A9+%D9%87%D8%B0%D9%87+%D8%A7%D9%84%D8%AE%D8%B7%D9%88%D8%A9+%D9%87%D9%8A%3A+d%28NOX2%29%2Fdt+%3D+K1%5BNO%5D+%5BX2%5D+%D8%AD%D9%8A%D8%AB+K1+%D9%87%D9%8A+%D8%AB%D8%A7%D8%A8%D8%AA+%D8%A7%D9%84%D8%B3%D8%B1%D8%B9%D8%A9+%D9%84%D9%87%D8%B0%D9%87+%D8%A7%D9%84%D8%AE%D8%B7%D9%88%D8%A9+%D9%8A%D8%AA%D8%A8%D8%B9+%D8%B0%D9%84%D9%83+%D8%A7%D9%84%D8%AA%D9%81%D8%A7%D8%B9%D9%84+%D8%A7%D9%84%D8%A8%D8%B7%D8%A6+%D8%A7%D9%84%D8%A2%D8%AA%D9%8A%3A+NOX2+%2B+NO+2NOX+%D9%88%D9%85%D8%B9%D8%A7%D8%AF%D9%84%D8%A9+%D8%A7%D9%84%D8%B3%D8%B1%D8%B9%D8%A9+%D8%A7%D9%84%D9%86%D9%87%D8%A7%D8%A6%D9%8A%D8%A9+%D9%87%D9%8A%3A+R%3DK2%5BNO%5D%5BNOX2%5D+%3DK2K1%5BNO%5D2%5BX2%5D+%3D+K%5BNO%5D2+%5BX2%5D+%D9%88%D9%87%D8%B0%D9%87+%D8%A7%D9%84%D9%85%D9%8A%D9%83%D8%A7%D9%86%D9%8A%D9%83%D9%8A%D8%A9+%D8%AA%D8%A3%D8%AE%D8%B0+%D9%8A+%D8%A7%D9%84%D8%A7%D8%B9%D8%AA%D8%A8%D8%A7%D8%B1+%D9%85%D8%B9%D8%A7%D8%AF%D9%84%D8%A9+%D8%B3%D8%B1%D8%B9%D8%A9+%D8%A7%D9%84%D8%AA%D9%81%D8%A7%D8%B9%D9%84+%D9.jpg "وقد تم افتراض ميكانيكية بديلة لهذا النوع من التفاعلات كما يلي: يحدث التوازن التالي أولاً: NO + X2 NOX2 حيث X2 تمثل الأكسجين أو أي غاز آخر ، معادلة سرعة هذه الخطوة هي: d(NOX2)/dt = K1[NO] [X2] حيث K1 هي ثابت السرعة لهذه الخطوة يتبع ذلك التفاعل البطئ الآتي: NOX2 + NO 2NOX ومعادلة السرعة النهائية هي: R=K2[NO][NOX2] =K2K1[NO]2[X2] = K[NO]2 [X2] وهذه الميكانيكية تأخذ ي الاعتبار معادلة سرعة التفاعل ومن الواضح أن التوازن الذي تتبعه الخطوة البطيئة يمكن ان يكون كالتالي: 2NO N2O2 N2O2 + X2 2NOX")
135
يؤخذ أيضاً في الاعتبار في معادلة سرعة التفاعل الفرق بين فكرة التصادمات الثلاثية وهذه الافتراضات من الميكانيزم ليست كثيرة حيث أن NOX2 أو N2O2 يمكن النظر إليهما وكأنهما جزيئان متلامسان متصادمان وافتراض التوازن يوضح سالبيه معامل الحرارة لأنه في درجات الحرارة العالية تتفكك الجزيئات الثنائية NOX2 أو N2O2 وبالتالي كلما قل تركيزها انخفضت سرعة التفاعل أي أن طاقة التنشيط تكون سالبة. وعموما فان انخفاض قيمة ثابت سرعة تفاعل K مع درجة الحرارة قد يكون بسبب عامل التردد الذي يتناسب مع الأس الثالث لدرجة الحرارة المطلقة (T).
..")
136
التفاعلات الأحادية الجزيئية: نظرية التصادم لا شأن لها بالتفاعلات الأحادية المباشرة ولكنها تناقش الموضوع من خلال ميكانيكية ليندمان لأنه بمجرد حصول الجزئ على مقدار كاف من الطاقة نتيجة إصطدامه فإن المسألة تصبح: كيف يحسب ثابت التفاعل لتفكك الجزيئين المتصادمين وبالتالي يبدأ عمل نظرية التصادم. ناتج A* العالم سلاتر عالج الموضوع بطريقة أخرى فهو يفترض أنه إذا تحصل الجزئ نتيجة اصطدامه على كمية كبيرة من الطاقة فيكون له تردد عال ونتيجة لذلك فإن تردد الجزئيء يؤدي إلى تفككه. وعموماً فإن محاولة توضيح معادلة أرهينوس وتطبيقها على التفاعلات الأحادية تكون معقدة وصعبة جداً.

137
حدود نظرية التصادم 1- التفاعل بين ثلاثي أثيل أمين و يوديد الايثيل في الحالة الغازية يحدث بسرعة أبطأ من المتوقع فتكون P=10-8. تحلل بعض السكريات في الوسط الحامضي أسرع من المتوقع وتكون قيمة P في هذه الحالة أكبر من 109. من المثالين السابقين يتضح أن نظرية التصادم ليست هي النموذج الصحيح لتفسير هذه التفاعلات وعليه فإن المتناقضات الناتجة من نظرية التصادم يمكن أن تنسب إلى الآتي: التفاعلات ثنائية الجزيئية في الحالة الغازية تشتمل على جزيئات كبيرة وغالباً ما تكون قيم P أقل بكثير من الوحدة لذلك فإن المواد المتفاعلة عديدة الذرات تحتاج إلى توجيه خاص قبل أن تتفاعل فمثلاً في تفاعل البيوتاداين (1) مع اكرالدهيد حسب المعادلة: اكرالدهيد البيوتاداين (1)
 مع اكرالدهيد حسب المعادلة: اكرالدهيد. البيوتاداين (1)")
138
يكون فيه قيمة عامل التردد أو ثابت أرهينيوس مساوية 610 لتر مول-1 ثانية-1 وهي قيمة صغيرة. أي أن وجود مجموعات كبيرة أو تركب خاص قد تصف وضعاً غير مقبول لتصادم نشط غير مميز. 2- يمكن أن تحدث التفاعلات في سلسلة من ميكانيكية التفاعل وليس في خطوة واحدة حيث يكون جزئ المواد الناتجة قادر على تفاعلات أخرى مما يسبب ارتفاع في قيمة معامل الاحتمال (P). 3- إذا حــــدث التـــفاعل على ســطح محفز فلا يمكن تطبيق نظرية التصادم لأن سرعة التفاعل لا تعتمد هنا على عدد التصادمات. 4- فترة التصادم هي مظهر آخر مهمل في نظرية التصادم البسيط للكرات المصمتة وتصور التصادم اللحظي ثنائي الجزئيء لا يدخل في اعتباره القوى ذات المدى القصير للتداخل بين الجزيئات المتفاعلة وعلى أي حالة فإنه في درجة الحرارة العادية يقدر الوقت المناسب الذي تكون خلاله هذه القوى مناسبة هو ثانية وهو نفس مقدار زمن دورة ذبذبة الجزيء
. 3- إذا حــــدث التـــفاعل على ســطح محفز فلا يمكن تطبيق نظرية التصادم لأن سرعة التفاعل لا تعتمد هنا على عدد التصادمات. 4- فترة التصادم هي مظهر آخر مهمل في نظرية التصادم البسيط للكرات المصمتة وتصور التصادم اللحظي ثنائي الجزئيء لا يدخل في اعتباره القوى ذات المدى القصير للتداخل بين الجزيئات المتفاعلة وعلى أي حالة فإنه في درجة الحرارة العادية يقدر الوقت المناسب الذي تكون خلاله هذه القوى مناسبة هو ثانية وهو نفس مقدار زمن دورة ذبذبة الجزيء .")
139
ثانياً : نظرية سرعة التفاعلات المطلقة: الأساس في فرضية نظرية سرعة التفاعلات المطلقة هو أن المواد المتفاعلة دائماً في اتزان مع المركب النشط والاتزان المفترض هنا يختلف عن الاتزان المطبق في الديناميكا الحرارية بالنسبة إلى التفاعلات العكسية. ويقصد بالمركب النشط ذلك الشكل للذرات الذي يطابق من ناحية الطاقة قمة حاجز الطاقة الذي يفصل بين المواد المتفاعلة والمواد الناتجة، كما هو موضح في الشكل التالي (5): يبين الشكل (5): الاختلاف في الطاقة عند تحول المواد المتفاعلة إلى ناتجة الاتزان المفترض يمكن كتابته كالتالي: A+B M≠ وعليه يكون ثابت الاتزان لهذا التفاعل هو:
: يبين الشكل (5): الاختلاف في الطاقة عند تحول المواد المتفاعلة إلى ناتجة الاتزان المفترض يمكن كتابته كالتالي: A+B M≠ وعليه يكون ثابت الاتزان لهذا التفاعل هو: .")
140
وعليه يكون ثابت الاتزان لهذا التفاعل هو: K=C≠/CA
وعليه يكون ثابت الاتزان لهذا التفاعل هو: K=C≠/CA.CB (67) تركيز المركب النشط C≠ = K≠.CA.CB بمعرفة تركيز المركب النشط يمكن تحليل المسألة لحساب سرعة التي تتفكك بـها هذه المركبات إلى مواد ناتجة أي أن تبدأ بحساب سرعة التفاعل كالتالي: نواتج M≠ من تعريف المركب النشط بأنه الشكل للذرات الذي يطابق من ناحية الطاقة قمة حاجز الطاقة، أي أنه عبارة عن مجموعة من الذرات لا تختلف عن غيرها إلا في أنها تحتوي على اهتزاز خاص بواسطته تكون غير مستقرة، هذا الاهتزاز يؤدي إلى تفكك المركب النشط ليعطي المواد الناتجة، فإذا كان تردد الاهتزاز هو ν فتكون سرعة تكوين الناتج كالتالي: جزئ/سم3/ثانية. السرعة R = ν C≠
 تركيز المركب النشط C≠ = K≠.CA.CB بمعرفة تركيز المركب النشط يمكن تحليل المسألة لحساب سرعة التي تتفكك بـها هذه المركبات إلى مواد ناتجة أي أن تبدأ بحساب سرعة التفاعل كالتالي: نواتج M≠ من تعريف المركب النشط بأنه الشكل للذرات الذي يطابق من ناحية الطاقة قمة حاجز الطاقة، أي أنه عبارة عن مجموعة من الذرات لا تختلف عن غيرها إلا في أنها تحتوي على اهتزاز خاص بواسطته تكون غير مستقرة، هذا الاهتزاز يؤدي إلى تفكك المركب النشط ليعطي المواد الناتجة، فإذا كان تردد الاهتزاز هو ν فتكون سرعة تكوين الناتج كالتالي: جزئ/سم3/ثانية. السرعة R = ν C≠")
141
باستخدام قيمة C≠ من المعادلة (67) نجد أن: R = ν K≠ CA CB (68) ولكن سرعة التفاعل الأولي بين: نواتج A + B هي كالتالي: R = K CA.CB (69) من المعادلتين (68) ، (69) ينتج أن: (ثابت سرعة التفاعل) K = ν K≠ (70) من الخطوات السابقة يتضح أن معادلة (70) غير مقيدة باختيار متفاعلين ولكنها صالحة لأي تفاعل أولي.
 نجد أن: R = ν K≠ CA CB (68) ولكن سرعة التفاعل الأولي بين: نواتج A + B هي كالتالي: R = K CA.CB (69) من المعادلتين (68) ، (69) ينتج أن: (ثابت سرعة التفاعل) K = ν K≠ (70) من الخطوات السابقة يتضح أن معادلة (70) غير مقيدة باختيار متفاعلين ولكنها صالحة لأي تفاعل أولي.")
142
قيمة تردد الإهــتزاز ν وثابت الاتزان للمركب K≠ يمكن حسابها إذا استخدمت حدود دالة فصل الجزئ في وحدة الحجم (f-) للتعبير عن ثابت الاتزان K≠ أي أن: K≠ = f-≠/f-A f-B (71) حيث دالة فصل الجزيء يمكن التعبير عنها بالقيمة الآتية: f- = fe-Є0/KT (72) حيث أن f- هي دالة فصل الجزئيء مقدرة باستخدام الطاقات بالنسبة إلى قيمة طاقة نقطة الصفر للجزيء وعليه فإن المعادلة (71) تصبح كالتالي: K≠ = f≠/fA fB e-(Єo≠ -ЄoA- ЄoB/kT) = f≠/fA fB eEo/RT (73)
 للتعبير عن ثابت الاتزان K≠ أي أن: K≠ = f-≠/f-A f-B (71) حيث دالة فصل الجزيء يمكن التعبير عنها بالقيمة الآتية: f- = fe-Є0/KT (72) حيث أن f- هي دالة فصل الجزئيء مقدرة باستخدام الطاقات بالنسبة إلى قيمة طاقة نقطة الصفر للجزيء وعليه فإن المعادلة (71) تصبح كالتالي: K≠ = f≠/fA fB e-(Єo≠ -ЄoA- ЄoB/kT) = f≠/fA fB eEo/RT (73)")
143
حيث تعرف طاقة التنشيط Eo بالفرق في طاقة نقطة الصفر بين المركب النشط والمواد المتفاعلة أي: Eo=No(Єo≠-ЄoA-ЄoB) دالة الفصل يمكن كتابتها كناتج لدوال الفصل بالنسبة إلى التنقل والدوران والاهتزاز Translation Rotation and Vibration بالإضافة إلى الاهتزاز الخاص الذي يؤدي إلى تفكك المركب النشط ليعطي المواد الناتجة وكذلك العامل من دالة اهتزاز الفصل الناتج من f≠لنفرض أن: f≠ = fv f≠ (74) حيث f≠ تمثل الباقي من f≠ بعد عمل fv ، ولكن fv قيمتها تعطي كالتالي: fv= kT/hν ehv/kT
 دالة الفصل يمكن كتابتها كناتج لدوال الفصل بالنسبة إلى التنقل والدوران والاهتزاز Translation Rotation and Vibration بالإضافة إلى الاهتزاز الخاص الذي يؤدي إلى تفكك المركب النشط ليعطي المواد الناتجة وكذلك العامل من دالة اهتزاز الفصل الناتج من f≠لنفرض أن: f≠ = fv f≠ (74) حيث f≠ تمثل الباقي من f≠ بعد عمل fv ، ولكن fv قيمتها تعطي كالتالي: fv= kT/hν ehv/kT")
144
فإذا كانت v صغيرة وكانت 1>> hv/kT فإن المقدار e-hv/kT يقارب العدد صفر ومنه فأن: fv = kT/hν و تؤول المعادلة (74) إلى: f≠ = KT/hν f≠ (75) وبالتعويض عن قيمة f≠ الناتجة في المعادلة (73) نحصل على: K≠ = kT/hν (f≠/fA fB) e-Eo/RT (76) بالتعويض في المعادلة (70) بقيمة K≠ يتم الحصول على: K = kT/h (f≠/fA fB) e-Eo/RT (77)
 إلى: f≠ = KT/hν f≠ (75) وبالتعويض عن قيمة f≠ الناتجة في المعادلة (73) نحصل على: K≠ = kT/hν (f≠/fA fB) e-Eo/RT (76) بالتعويض في المعادلة (70) بقيمة K≠ يتم الحصول على: K = kT/h (f≠/fA fB) e-Eo/RT (77)")
145
بمقارنة هذه المعادلة مع معادلة أرهينيوس فأن عامل التردد A (عامل أرهينوس) يمثل المقدار:
A = kT/h (f≠/ fA fB) (78) المعادلة (78) مهمة لأن دالة الفصل تعتمد على درجات الانطلاق للانتقال وللمركب النشط على درجة الانطلاق الداخلية أيضاً. لحساب ثابت سرعة التفاعل من المعادلة (77) يتطلب أمرين هما: 1- صفات المركب النشط يجب أن تكون كاملة حتى يمكن حساب قيمة f≠ وهذا يتطلب معرفة حجم الجزيئات وشكلها بحيث يمكن حساب عزم القصور الذاتي، كما أن حساب تردد الاهتزازات يمكن أن يتم بواسطة ميكانيكا الكم، ولو أنه معقد. 2- يجب معرفة قيمة Eo وحسابها بواسطة ميكانيكا الكم معقد للغاية إلا إذا استعمل التقريب بدرجة فعالة وهذه الطريقة استخدمت بالتفصيل لعدد من التفاعلات ولاسيما التي تشمل ذرات الهدروجين وجزيئات الهيدروجين وبالرغم من اشتمال الطريقة على التقريب فإن النتائج جيدة جداً وليس من الصعب نسبياً الحصول على تقدير تقريبي لدرجة كمية عامل التردد باستعمال المعادلة (78).
 (78) المعادلة (78) مهمة لأن دالة الفصل تعتمد على درجات الانطلاق للانتقال وللمركب النشط على درجة الانطلاق الداخلية أيضاً. لحساب ثابت سرعة التفاعل من المعادلة (77) يتطلب أمرين هما: 1- صفات المركب النشط يجب أن تكون كاملة حتى يمكن حساب قيمة f≠ وهذا يتطلب معرفة حجم الجزيئات وشكلها بحيث يمكن حساب عزم القصور الذاتي، كما أن حساب تردد الاهتزازات يمكن أن يتم بواسطة ميكانيكا الكم، ولو أنه معقد. 2- يجب معرفة قيمة Eo وحسابها بواسطة ميكانيكا الكم معقد للغاية إلا إذا استعمل التقريب بدرجة فعالة وهذه الطريقة استخدمت بالتفصيل لعدد من التفاعلات ولاسيما التي تشمل ذرات الهدروجين وجزيئات الهيدروجين وبالرغم من اشتمال الطريقة على التقريب فإن النتائج جيدة. جداً وليس من الصعب نسبياً الحصول على تقدير تقريبي لدرجة كمية عامل التردد باستعمال المعادلة (78).")
146
جزئية عديدة الذرات على شكل خطي جزئية عديدة الذرات غير خطية
يوضح الجدول الآتي طريقة حساب درجات الحرية للذرات أو الجزيئات. درجات الحرية الذرة الأحادية ثنائي الذرة جزئية عديدة الذرات على شكل خطي جزئية عديدة الذرات غير خطية العدد الكلي لدرجات الحرية (3N) 3 6 3N درجات الحرية للإنتقال ft درجات الحرية للدوران fr - 2 درجات الحرية للإهتزاز fv 1 (5- 3N) (6-3N) حيث (N) تساوي عدد الذرات المكونة للجزئ.
 N. درجات الحرية للإنتقال ft. درجات الحرية للدوران fr درجات الحرية للإهتزاز fv. 1. (5- 3N) (6-3N) حيث (N) تساوي عدد الذرات المكونة للجزئ.")
147
معادلة عامل التردد تكون كالتالي: A = kT/h f≠/ fH2 fI2
مثال: تفاعل اليود مع الهيدروجين: H I HI معادلة عامل التردد تكون كالتالي: A = kT/h f≠/ fH2 fI2 دالة الفصل fH2 يمكن التعبير عنها كناتج لدوال الفصل التالية: درجات الحرية بالنسبة إلى الانتقال (f3t). درجتي الحرية بالنسبة إلى الدوران (f2r) درجة الحرية بالنسـبة إلى الإهــتزاز (fv) fI2 = fH2 = ft3.fr2.fv حيث استعملت نفس الدالة بالنسبة لليود ( نفس عدد الذرات في الجزيئين ) وذلك حتى يمكن حساب أس الكمية، المركب (HI)2 له ثلاث درجات حرية بالنسبة إلى الانتقال ومثلها بالنسبة إلى الدوران وست درجات حرية بالنسبة إلى الإهتزاز، كما في المعادلة الآتية: f≠ = ft3. fr3. fv6 باستعمال هذه القيم في معادلة عامل التردد يتم الحصول على: A =kT/h (ft3 fr3 fv6/(ft3 fr2 fv ft3 fr2 fv ) = kT/h (fv4/ ft3 fr)
. درجتي الحرية بالنسبة إلى الدوران (f2r) درجة الحرية بالنسـبة إلى الإهــتزاز (fv) fI2 = fH2 = ft3.fr2.fv. حيث استعملت نفس الدالة بالنسبة لليود ( نفس عدد الذرات في الجزيئين ) وذلك حتى يمكن حساب أس الكمية، المركب (HI)2 له ثلاث درجات حرية بالنسبة إلى الانتقال ومثلها بالنسبة إلى الدوران وست درجات حرية بالنسبة إلى الإهتزاز، كما في المعادلة الآتية: f≠ = ft3. fr3. fv6. باستعمال هذه القيم في معادلة عامل التردد يتم الحصول على: A =kT/h (ft3 fr3 fv6/(ft3 fr2 fv ft3 fr2 fv ) = kT/h (fv4/ ft3 fr)")
148
في درجات الحرارة العادية أس الكمية لهذه المقادير في الحدود التالية: ft = 108 , fr = 10, fv 1, kT/h 1013 بالتعويض بهذه القيم في المعادلة السابقة ينتج أن: A = 1013/ (108)3 10 = cm2/atom/sec لتحويل هذه القيمة من سم3/جزئ/ثانية إلى لتر/وزن جزئ/ثانية يضرب الناتج في المقدار No/1000. A = 10-12x 6x1023/1000 = 109L/mol/Sec هذه القيمة لعامل التردد (A) تتفق بالتقريب مع قيمته التي وجدت للتفاعلات الثنائية وهي دائماً بين 910 ، 1110 فإذا استخدمت القيم التقريبية المستعملة لدوال الفصل فإن اتفاق النتائج يكون جيد جداً.
 تتفق بالتقريب مع قيمته التي وجدت للتفاعلات الثنائية وهي دائماً بين 910 ، 1110 فإذا استخدمت القيم التقريبية المستعملة لدوال الفصل فإن اتفاق النتائج يكون جيد جداً.")
149
علاقة الطاقة الحرة وأنتروبي التنشيط بثابت سرعة التفاعل: يقصد بالانتروبي: محددة التغيير الخاصة بالنظام وهي الوحدة أو المقياس الذي تقاس به لا إتاحية الطاقة في مجموعة ديناميكية حرارية أو مجموعة طاقة حرارية ، ومعنى هذا أنه إذا أتيح لمجموعة فيزيقية توزيع طاقتها في معزل عن المؤثرات الخارجية فإن الانتروبي يزداد مع نقص الطاقة المتاحة لمجموعة فيزيقية توزيع طاقتها في معزل عن المؤثرات الخارجية فإن الانتروبي يزداد مع نقص الطاقة المتاحة. حسب معادلة السرعة في نظرية السرعات المطلقة فإن: K= (kT/h) (f≠ / fA fB ( e-Eo/RT ولكن من تعريف قيمة K≠ فإن: K≠ = (f≠/ fA fB )e-Eo/RT حيث K≠ ثابت الاتزان للنظام ككل في الحالة النشطة: K= kT/h K≠ (83)
 (f≠ / fA fB ( e-Eo/RT ولكن من تعريف قيمة K≠ فإن: K≠ = (f≠/ fA fB )e-Eo/RT حيث K≠ ثابت الاتزان للنظام ككل في الحالة النشطة: K= kT/h K≠ (83) .")
150
ولكن: K≠ = e-∆G≠/RT حيث ∆G≠ طاقة التنشيط الحرة القياسية ولكن من القانون الثاني للديناميكا الحرارية فان: ∆G≠ = ∆H≠ - T∆S≠ حيث ∆S≠ قيمة التغيير في أنتروبي للتنشيط، ∆H≠ هي المحتوى الحرارية للتنشيط. K≠ = e-(∆H*-T∆S≠ / RT) K≠ =∆S≠/RT e-∆H≠/RT (84) بالتعويض في المعادلة (83) عن قيمة K بما يساويها من المعادلة (84) ينتج أن: K= kT/h e∆S≠/R e-∆H≠/RT (85) وهذه المعادلة تشبه معادلة أرهينوس باستثناء ظهور ∆H≠ بدلاً من E*، وعليه فإن عامل التردد A يكون: K= kT/h e∆S≠/R سالبية أنتروبي التنشيط تؤدي إلى قيمة منخفضة لعامل التردد بينما إذا كان أنتروبي التنشيط موجباً فسوف يرفع من قيمة عامل التردد . A
 K≠ =∆S≠/RT e-∆H≠/RT (84) بالتعويض في المعادلة (83) عن قيمة K بما يساويها من المعادلة (84) ينتج أن: K= kT/h e∆S≠/R e-∆H≠/RT (85) وهذه المعادلة تشبه معادلة أرهينوس باستثناء ظهور ∆H≠ بدلاً من E*، وعليه فإن عامل التردد A يكون: K= kT/h e∆S≠/R. سالبية أنتروبي التنشيط تؤدي إلى قيمة منخفضة لعامل التردد بينما إذا كان أنتروبي التنشيط موجباً فسوف يرفع من قيمة عامل التردد . A.")
151
كذلك من نظرية التصادم. A = PZ- PZ- = kT/h e(∆S≠/R) تردد الاصطدام Z- يمكن حسابه بسهولة ومن معرفة قيمةP يمكن إيجاد قيمة أنتروبي التنشيط ∆S≠ إذا كان المركب النشط يشبه المواد الناتجة أكثر من المواد المتفاعلة فإن أنتروبي التنشيط يمكن بالتقريب أن يساوي أنتروبي التفاعل الكلي Sº∆ ، القيم ∆S≠ ، ∆Sº يمكن أن توازي إحداهما الأخرى في حالات كثيرة ولو أنه من النادر أن تكونا متساويتين.
 تردد الاصطدام Z- يمكن حسابه بسهولة ومن معرفة قيمةP يمكن إيجاد قيمة أنتروبي التنشيط ∆S≠ إذا كان المركب النشط يشبه المواد الناتجة أكثر من المواد المتفاعلة فإن أنتروبي التنشيط يمكن بالتقريب أن يساوي أنتروبي التفاعل الكلي Sº∆ ، القيم ∆S≠ ، ∆Sº يمكن أن توازي إحداهما الأخرى في حالات كثيرة ولو أنه من النادر أن تكونا متساويتين.")
152
التفاعلات في المحلول ثابت التفاعل لتفاعل ما في الحالة السائلة غالباً ما يكون بالتقريب مساوياً للتفاعل نفسه في الحالة الغازية، أما إذا كان ثابت التفاعل في الحالة السائلة يختلف كثيراً عنه للتفاعل نفسه في الحالة الغازية فإن هذا يدل على أن تفاعلاً قوياً يتم بين المذيب والجزيئات المتفاعلة أو بين المذيب والمركب النشط أما سبب المساواة بالتقريب بين ثابت التفاعل في الحالة السائلة والحالة الغازية للتفاعل نفسه فيمكن توضيحه بسهولة باستخدام نظرية التصادم. إذا افترض أن تفاعلاً تطلب التصادم بين جزيئين في الحالة الغازية فإن : A + A → ناتج عدد مرات التصادم في كل سنتيمتر مكعب في الثانية هو كالتالي: ZII = Zn2A نفرض أن جزيئاً غريباً B وجد في وسط التفاعل فيكون هناك تصادم بين B,A وتصادم بين B,B وهذه الحقيقة لا تغير عدد مرات التصادم A,A والتي تعطي بالمعادلة السابقة نفسها.

153
وعلى هذا حتى إذا شغلت المسافات الداخلية بين جزيئات الغاز بجزيئات غريبة مثل B فإن ثابت التفاعل يظل دون تغيير على هذا الأساس يكون ثابت التفاعل في الحالة السائلة تقريبًا له نفس القيمة للحالة الغازية. هذا الفرض يكون صحيحاً للمحاليل المثالية المعقولة فقط لأن عدم المثالية في المحاليل يدخل فيها تأثير الإذابة. فإذا فرض التفاعل الآتي في الحالة السائلة: A + B → M≠ ناتج بتطبيق نظريات سرعات التفاعل المطلقة: K≠ = a≠/aA aB = v≠c≠ /νAcA vBcB حيث a≠ فعالية المركب النشط aA.aB فعالية المواد المتفاعلة B , A وكل من vA,vB,v≠ التنشيط (الفعالية) لكل من المركب النشط والمواد المتفاعلة B,A على التوالي. C≠ = K≠(νA νB cA cB /ν≠) R = vC≠ = vK≠ (vA.vB.cA.cB/v≠)
 لكل من المركب النشط والمواد المتفاعلة B,A على التوالي. C≠ = K≠(νA νB cA cB /ν≠) R = vC≠ = vK≠ (vA.vB.cA.cB/v≠) .")
154
ولكن من قانون الدرجة الثانية: R = KCA. CB K = vK≠ (vA
ولكن من قانون الدرجة الثانية: R = KCA.CB K = vK≠ (vA.vB/v≠) ولكن: K≠ = (kT/ vh) K≠ حيث K≠ هي ثابت الإتزان للنظام K = (kT/h) K≠ (vA.vB/v≠) وهذه المعادلة هي التي تعبر عن ثابت التفاعل في الحالة السائلة. لموازنة معادلة سرعة التفاعل في المحاليل بالمعادلة في الحالة الغازية نفترض الحالة التي تكون فيها v=1 وهي التي تمثل الغاز المثالي وبالتالي تؤول المعادلة (86) إلى الصورة: Kg= (kT/h ) K≠ حيث Kg تمثل ثابت سرعة التفاعل في الحالة الغازية، وبالتعويض بهذه القيمة في المعادلة (86) يتم الحصول على:
 ولكن: K≠ = (kT/ vh) K≠ حيث K≠ هي ثابت الإتزان للنظام K = (kT/h) K≠ (vA.vB/v≠) وهذه المعادلة هي التي تعبر عن ثابت التفاعل في الحالة السائلة. لموازنة معادلة سرعة التفاعل في المحاليل بالمعادلة في الحالة الغازية نفترض الحالة التي تكون فيها v=1 وهي التي تمثل الغاز المثالي وبالتالي تؤول المعادلة (86) إلى الصورة: Kg= (kT/h ) K≠ حيث Kg تمثل ثابت سرعة التفاعل في الحالة الغازية، وبالتعويض بهذه القيمة في المعادلة (86) يتم الحصول على:")
155
K = Kg(vA. vB/v≠) (87) اختلاف قيمة ثابت سرعة التفاعل في الحال السائلة عن قيمته في الحالة الغازية يعتمد على قيمة معامل التنشيط v، فإذا كان المذيب يخفض قيمة الطاقة الحرة للمواد المتفاعلة أكثر من خفضه لقيمة الطاقة الحرة للمركب النشط (وهذا يعني أن المواد المتفاعلة تذوب بشدة) فإن vB,vA تكون صغيرة بينما تكون v≠ بالمقارنة كبيرة وبذلك يكون ثابت سرعة التفاعل في الحالة السائلة أصغر منه في الحالة الغازية. أما إذا كان المركب النشط هو الذي أذيب بشدة أكثر من المواد المتفاعلة فأن ثابت سرعة التفاعل في هذه الحالة يكون اكبر في الحالة السائلة منه في الحالة الغازية. إذا كان التفاعل من التفاعلات التي لا تحدث في الحالة الغازية فإنه من المفيد جداً اختيار المحلول المخفف إلى مالانهاية لتكون حالة المرجع الذي معامل التنشيط له هو الوحدة (v=1) فإذا كان ثابت التفاعل في هذه الحالة المخفضة لما لا نهاية يعبر عنه بالرمز K0 حيث أن (v=1) فإن معادلة التفاعل تؤول إلى: K0 = (kT/h )K≠ K =K0(vA.vB/v≠) (88)
 فإن vB,vA تكون صغيرة بينما تكون v≠ بالمقارنة كبيرة وبذلك يكون ثابت سرعة التفاعل في الحالة السائلة أصغر منه في الحالة الغازية. أما إذا كان المركب النشط هو الذي أذيب بشدة أكثر من المواد المتفاعلة فأن ثابت سرعة التفاعل في هذه الحالة يكون اكبر في الحالة السائلة منه في الحالة الغازية. إذا كان التفاعل من التفاعلات التي لا تحدث في الحالة الغازية فإنه من المفيد جداً اختيار المحلول المخفف إلى مالانهاية لتكون حالة المرجع الذي معامل التنشيط له هو الوحدة (v=1) فإذا كان ثابت التفاعل في هذه الحالة المخفضة لما لا نهاية يعبر عنه بالرمز K0 حيث أن (v=1) فإن معادلة التفاعل تؤول إلى: K0 = (kT/h )K≠ K =K0(vA.vB/v≠)0 (88)")
156
والصفر المذيل في قوس معامل التنشيط يدل على حالة المرجع التي هي المحلول المخفف إلى مالا نهاية ، ولكن عند استخدام المحاليل المخفضة تصبح المعادلة: K = Ko(vA.vB/v≠) (89) المعادلة (89) اشتقت أولاً بواسطة برونشتيد وبيرم قبل تطبيق نظرية التفاعلات المطلق على التفاعلات الأيونية، بأخذ لوغاريتمات الطرفين لطرفي المعادلة نجد أن: log K = logK0 + logvA+ logvB - logv≠ (90) ولكن من معادلة ديبى- هيكل لمعامل التنشيط حيث: log vi = - 0.5Zi2√I حيث Z هي شحنة الأيون I , i هي القوة الأيونية وتعطي قيمتها من المعادلة: I = 1/2 ∑i Zi2 Ci بالتعويض عن قيمة معامل الفعالية في المعادلة (90) ينتج أن: log K = logK0 – 0.5√I [ZA2 + ZB2-(ZA+ZB)2] حيث: ZAB = (ZA + ZB)
![والصفر المذيل في قوس معامل التنشيط يدل على حالة المرجع التي هي المحلول المخفف إلى مالا نهاية ، ولكن عند استخدام المحاليل المخفضة تصبح المعادلة: K = Ko(vA.vB/v≠) (89) المعادلة (89) اشتقت أولاً بواسطة برونشتيد وبيرم قبل تطبيق نظرية التفاعلات المطلق على التفاعلات الأيونية، بأخذ لوغاريتمات الطرفين لطرفي المعادلة نجد أن: log K = logK0 + logvA+ logvB - logv≠ (90) ولكن من معادلة ديبى- هيكل لمعامل التنشيط حيث: log vi = - 0.5Zi2√I حيث Z هي شحنة الأيون I , i هي القوة الأيونية وتعطي قيمتها من المعادلة: I = 1/2 ∑i Zi2 Ci بالتعويض عن قيمة معامل الفعالية في المعادلة (90) ينتج أن: log K = logK0 – 0.5√I [ZA2 + ZB2-(ZA+ZB)2] حيث: ZAB = (ZA + ZB)](http://slideplayer.ae/slide/15551337/88/images/156/%D9%88%D8%A7%D9%84%D8%B5%D9%81%D8%B1+%D8%A7%D9%84%D9%85%D8%B0%D9%8A%D9%84+%D9%81%D9%8A+%D9%82%D9%88%D8%B3+%D9%85%D8%B9%D8%A7%D9%85%D9%84+%D8%A7%D9%84%D8%AA%D9%86%D8%B4%D9%8A%D8%B7+%D9%8A%D8%AF%D9%84+%D8%B9%D9%84%D9%89+%D8%AD%D8%A7%D9%84%D8%A9+%D8%A7%D9%84%D9%85%D8%B1%D8%AC%D8%B9+%D8%A7%D9%84%D8%AA%D9%8A+%D9%87%D9%8A+%D8%A7%D9%84%D9%85%D8%AD%D9%84%D9%88%D9%84+%D8%A7%D9%84%D9%85%D8%AE%D9%81%D9%81+%D8%A5%D9%84%D9%89+%D9%85%D8%A7%D9%84%D8%A7+%D9%86%D9%87%D8%A7%D9%8A%D8%A9+%D8%8C+%D9%88%D9%84%D9%83%D9%86+%D8%B9%D9%86%D8%AF+%D8%A7%D8%B3%D8%AA%D8%AE%D8%AF%D8%A7%D9%85+%D8%A7%D9%84%D9%85%D8%AD%D8%A7%D9%84%D9%8A%D9%84+%D8%A7%D9%84%D9%85%D8%AE%D9%81%D8%B6%D8%A9+%D8%AA%D8%B5%D8%A8%D8%AD+%D8%A7%D9%84%D9%85%D8%B9%D8%A7%D8%AF%D9%84%D8%A9%3A+K+%3D+Ko%28vA.vB%2Fv%E2%89%A0%29+%2889%29+%D8%A7%D9%84%D9%85%D8%B9%D8%A7%D8%AF%D9%84%D8%A9+%2889%29+%D8%A7%D8%B4%D8%AA%D9%82%D8%AA+%D8%A3%D9%88%D9%84%D8%A7%D9%8B+%D8%A8%D9%88%D8%A7%D8%B3%D8%B7%D8%A9+%D8%A8%D8%B1%D9%88%D9%86%D8%B4%D8%AA%D9%8A%D8%AF+%D9%88%D8%A8%D9%8A%D8%B1%D9%85+%D9%82%D8%A8%D9%84+%D8%AA%D8%B7%D8%A8%D9%8A%D9%82+%D9%86%D8%B8%D8%B1%D9%8A%D8%A9+%D8%A7%D9%84%D8%AA%D9%81%D8%A7%D8%B9%D9%84%D8%A7%D8%AA+%D8%A7%D9%84%D9%85%D8%B7%D9%84%D9%82+%D8%B9%D9%84%D9%89+%D8%A7%D9%84%D8%AA%D9%81%D8%A7%D8%B9%D9%84%D8%A7%D8%AA+%D8%A7%D9%84%D8%A3%D9%8A%D9%88%D9%86%D9%8A%D8%A9%D8%8C+%D8%A8%D8%A3%D8%AE%D8%B0+%D9%84%D9%88%D8%BA%D8%A7%D8%B1%D9%8A%D8%AA%D9%85%D8%A7%D8%AA+%D8%A7%D9%84%D8%B7%D8%B1%D9%81%D9%8A%D9%86+%D9%84%D8%B7%D8%B1%D9%81%D9%8A+%D8%A7%D9%84%D9%85%D8%B9%D8%A7%D8%AF%D9%84%D8%A9+%D9%86%D8%AC%D8%AF+%D8%A3%D9%86%3A+log+K+%3D+logK0+%2B+logvA%2B+logvB+-+logv%E2%89%A0+%2890%29+%D9%88%D9%84%D9%83%D9%86.jpg "والصفر المذيل في قوس معامل التنشيط يدل على حالة المرجع التي هي المحلول المخفف إلى مالا نهاية ، ولكن عند استخدام المحاليل المخفضة تصبح المعادلة: K = Ko(vA.vB/v≠) (89) المعادلة (89) اشتقت أولاً بواسطة برونشتيد وبيرم قبل تطبيق نظرية التفاعلات المطلق على التفاعلات الأيونية، بأخذ لوغاريتمات الطرفين لطرفي المعادلة نجد أن: log K = logK0 + logvA+ logvB - logv≠ (90) ولكن من معادلة ديبى- هيكل لمعامل التنشيط حيث: log vi = - 0.5Zi2√I حيث Z هي شحنة الأيون I , i هي القوة الأيونية وتعطي قيمتها من المعادلة: I = 1/2 ∑i Zi2 Ci بالتعويض عن قيمة معامل الفعالية في المعادلة (90) ينتج أن: log K = logK0 – 0.5√I [ZA2 + ZB2-(ZA+ZB)2] حيث: ZAB = (ZA + ZB)")
157
ومنه: logK = logK0 - 05√I [-2ZA. ZB] = logK0 + ZA. ZB√I logK/K0 = ZA
ومنه: logK = logK0 - 05√I [-2ZA.ZB] = logK0 + ZA.ZB√I logK/K0 = ZA.ZB√I (91) برسم العلاقة بين( log(K/K0 والجذر التربيعي لقوة الأيونات (I) فإنه يتم الحصول في المحاليل المخففة على خط مستقيم ميله يساوي ZA ZB. كــما توضح هذه الـعلاقة أن قيمة ثابت سرعة التفاعل تعتمد علــى قيمة وإشارة الأيونات الموجودة في المحلول وعليه فهناك ثلاثة أنواع من السلوك يمكن تمييزها: 1- عندما تكون ZA ZB موجبة فإن قيمة ثابت سرعة التفاعل K سوف تزداد مع زيادة القوة الأيونية للأيونات الموجودة بالمحلول. 2- عندما تكون ZA ZB سالبة فإن قيمة ثابت سرعة التفاعل K سوف تقل مع زيادة القوة الأيونية (I). 3-عندما تكون ZA ZB = صفر في المحاليل المخففة لانهائياً فإن قيمة (K) لا تتأثر بتركيز الأيونات.
![ومنه: logK = logK0 - 05√I [-2ZA. ZB] = logK0 + ZA. ZB√I logK/K0 = ZA](http://slideplayer.ae/slide/15551337/88/images/157/%D9%88%D9%85%D9%86%D9%87%3A+logK+%3D+logK0+-+05%E2%88%9AI+%5B-2ZA.+ZB%5D+%3D+logK0+%2B+ZA.+ZB%E2%88%9AI+logK%2FK0+%3D+ZA.jpg "ومنه: logK = logK0 - 05√I [-2ZA.ZB] = logK0 + ZA.ZB√I logK/K0 = ZA.ZB√I (91) برسم العلاقة بين( log(K/K0 والجذر التربيعي لقوة الأيونات (I) فإنه يتم الحصول في المحاليل المخففة على خط مستقيم ميله يساوي ZA ZB. كــما توضح هذه الـعلاقة أن قيمة ثابت سرعة التفاعل تعتمد علــى قيمة وإشارة الأيونات الموجودة في المحلول وعليه فهناك ثلاثة أنواع من السلوك يمكن تمييزها: 1- عندما تكون ZA ZB موجبة فإن قيمة ثابت سرعة التفاعل K سوف تزداد مع زيادة القوة الأيونية للأيونات الموجودة بالمحلول. 2- عندما تكون ZA ZB سالبة فإن قيمة ثابت سرعة التفاعل K سوف تقل مع زيادة القوة الأيونية (I). 3-عندما تكون ZA ZB = صفر في المحاليل المخففة لانهائياً فإن قيمة (K) لا تتأثر بتركيز الأيونات.")
158
المعادلة (91) تصف من وجهة نظر الكيمياء الحركية تأثير الملح أو بعبارة أبسط توضح تأثير الملح الأولي. الشكل التالي شكل (6) يوضح هذه المعادلة والاتفاق ناجح جداً ومقنع كما يتبين من الأمثلة التالية على هذه المعادلة: شكل(6): يوضح تأثير الملح الأولي
: يوضح تأثير الملح الأولي.")
159
[C8(NH3)5Br]2+ + Hg2+ S2O82- + I- NO2NCO2C2H5-+I- C12H22O11+OH-
ZAZB=4 [C8(NH3)5Br]2+ + Hg2+ I ZAZB=2 S2O82- + I- II ZAZB=1 NO2NCO2C2H5-+I- III ZAZB=0 C12H22O11+OH- IV ZAZB=-1 H2O2+H+ +Br- V ZAZB=-2 [Co(NH3)5Br]2+ +OH- VI التفسير الفيزيائي لهذا السلوك في حالات الشحنات المتشابهة والمختلفة هو أنه نتيجة الشحنة النسبية على المركب النشط، فإن قيمة معامل التنشيط تنخفض مع القيمة الاسية للرمز Z2، فإذا كانت جميع الأيونات لها شحنات تشابهه فإن المركب النشط تكون شحنته النسبية عالية إذا ماوزنت بأي واحدة من الشحنات الأخرى، وهذا يعني أن v≠ صغيرة جداً، والنسبة vA.vB/v≠ تكون عالية.
![[C8(NH3)5Br]2+ + Hg2+ S2O82- + I- NO2NCO2C2H5-+I- C12H22O11+OH-](http://slideplayer.ae/slide/15551337/88/images/159/%5BC8%28NH3%295Br%5D2%2B+%2B+Hg2%2B+S2O82-+%2B+I-+NO2NCO2C2H5-%2BI-+C12H22O11%2BOH-.jpg "ZAZB=4. [C8(NH3)5Br]2+ + Hg2+ I. ZAZB=2. S2O82- + I- II. ZAZB=1. NO2NCO2C2H5-+I- III. ZAZB=0. C12H22O11+OH- IV. ZAZB=-1. H2O2+H+ +Br- V. ZAZB=-2. [Co(NH3)5Br]2+ +OH- VI. التفسير الفيزيائي لهذا السلوك في حالات الشحنات المتشابهة والمختلفة هو أنه نتيجة الشحنة النسبية على المركب النشط، فإن قيمة معامل التنشيط تنخفض مع القيمة الاسية للرمز Z2، فإذا كانت جميع الأيونات لها شحنات تشابهه فإن المركب النشط تكون شحنته النسبية عالية إذا ماوزنت بأي واحدة من الشحنات الأخرى، وهذا يعني أن v≠ صغيرة جداً، والنسبة vA.vB/v≠ تكون عالية.")
160
1- التقليل من قيمة القوة الأيونية الحقيقة (I) في المحلول.
أما إذا كانت الأيونات لها شحنة مختلفة فإن الشحنة النسبية للمركب النشط تكون صغيرة إذا ماقورنت بأي واحدة من الشحنات الأخرى وهذا يعني أن v≠ اكبر من vA.vB/v≠ ومحصلة النتيجة أن النسبة vA,vB وكذلك سرعة التفاعل تكون صغيرة. وأخيراً إذا كانت إحدى المواد المتفاعلة غير مشحونة (B مثلاً متعادلة) فإن A والمركب النشط يكون لهما الشحنة نفسها والنسبة (vA/v≠) تساوي الوحدة ومستقلة عن √I عن وقيمة vB لاتتأثر بالتغير في قوة الأيونات (I) لأن B عبارة عن جزئ متعادل. عند تطبيق المعادلة (91) على المحاليل المركزة نسبياً لوحظ حدوث بعض الحيود وقد عزى ذلك إلى تكوين الأيون المزدوج في هذه المحاليل، وقد يكون الايون المزدوج المتكون مترابط بالكامل بروابط أيونية كما في حالة محلول 0.1 مولار نترات البوتاسيوم KNO3 حيث يكون حوالي 3% من أيونات الأيون المزدوج أو قد يتكون مركب كيميائي له تركيب محدد داخل المحلول مثل تكون متراكب CdI3 في محاليل 0.5 مولار حيث يشكل هذا المتراكب 24% من الأيونات. وعموماً فإن تكون الأيون المزدوج في المحاليل يؤثر على قيمة ثابت سرعة التفاعل بطرق عديدية منها الطريقتين الآتيتين: 1- التقليل من قيمة القوة الأيونية الحقيقة (I) في المحلول. 2- قد يحتوي الأيون المزدوج المتكون في تركيبه أحد الأيونات المتفاعلة أو كلاهما مما يقلل من التفاعل بين الأيونات المتفاعلة داخل المحلول.
 فإن A والمركب النشط يكون لهما الشحنة نفسها والنسبة (vA/v≠) تساوي الوحدة ومستقلة عن √I عن وقيمة vB لاتتأثر بالتغير في قوة الأيونات (I) لأن B عبارة عن جزئ متعادل. عند تطبيق المعادلة (91) على المحاليل المركزة نسبياً لوحظ حدوث بعض الحيود وقد عزى ذلك إلى تكوين الأيون المزدوج في هذه المحاليل، وقد يكون الايون المزدوج المتكون مترابط بالكامل بروابط أيونية كما في حالة محلول 0.1 مولار نترات البوتاسيوم KNO3 حيث يكون حوالي 3% من أيونات الأيون المزدوج أو قد يتكون مركب كيميائي له تركيب محدد داخل المحلول مثل تكون متراكب CdI3 في محاليل 0.5 مولار حيث يشكل هذا المتراكب 24% من الأيونات. وعموماً فإن تكون الأيون المزدوج في المحاليل يؤثر على قيمة ثابت سرعة التفاعل بطرق عديدية منها الطريقتين الآتيتين: 1- التقليل من قيمة القوة الأيونية الحقيقة (I) في المحلول. 2- قد يحتوي الأيون المزدوج المتكون في تركيبه أحد الأيونات المتفاعلة أو كلاهما مما يقلل من التفاعل بين الأيونات المتفاعلة داخل المحلول.")
161
مســــائل .اعتماد معدل التفاعل الأيوني على القوة الأيونية للتفاعل: [CoBr(NH3)5]+2 +OH- = [Co(NH3)5]+2 +Br- (1) أعطي كل من برونشتد J.R.Bronsted وليفنتون R.Livingston قيمة ثابت المعدل عند 15ºم بالمقدار: لتر.مول-1 . دقيقة K=91 مقاسه خلال التجربة عندما يكون التركيز الأصلي للأيون [CoBr(NH3)5]+2 (الموجود بشكل بروميد) 0.5x10-3M ، وكان تركيز ايون OH x10-3M = ، من هذه النتائج أحسبي: (أ). ثابت المعدل للتفاعل (1) إذا تم التفاعل عند التخفيف إلى مالا نهاية (ب) ثابت المعدل للتفاعل المذكور عند التراكيز المختلفة لكل من الأيونات المتفاعلة وفي وجود كميات مختلفة من كلوريد الصوديوم كما هو في الجدول التالي قارني النتائج مع المعلومات المحصل عليها عملياً بواسطة الباحثين السابقين (أنظري آخر حقل في الجدول). I=2.2x10-3 , log ko=2.052 , k0=113L.M-1min-1 [Co Br(NH3)5]+2 + OH- = [Co(NH3)5]+2 +Br-
5]+2 +OH- = [Co(NH3)5]+2 +Br- (1) أعطي كل من برونشتد J.R.Bronsted وليفنتون R.Livingston قيمة ثابت المعدل عند 15ºم بالمقدار: لتر.مول-1 . دقيقة K=91 مقاسه خلال التجربة عندما يكون التركيز الأصلي للأيون [CoBr(NH3)5]+2 (الموجود بشكل بروميد) 0.5x10-3M ، وكان تركيز ايون OH x10-3M = ، من هذه النتائج أحسبي: (أ). ثابت المعدل للتفاعل (1) إذا تم التفاعل عند التخفيف إلى مالا نهاية (ب) ثابت المعدل للتفاعل المذكور عند التراكيز المختلفة لكل من الأيونات المتفاعلة وفي وجود كميات مختلفة من كلوريد الصوديوم كما هو في الجدول التالي قارني النتائج مع المعلومات المحصل عليها عملياً بواسطة الباحثين السابقين (أنظري آخر حقل في الجدول). I=2.2x10-3 , log ko=2.052 , k0=113L.M-1min-1 [Co Br(NH3)5]+2 + OH- = [Co(NH3)5]+2 +Br-")
162
I =1/2[C1x 12 +C1x12] +1/2[CAx22 +2CAX12] +1/2[CBX(1)2 +CBX12]
A + B → C D I =1/2[C1x 12 +C1x12] +1/2[CAx CAX12] +1/2[CBX(1)2 +CBX12] =1/2[2C1 ] + 1/2[4CA+2CA] +1/2[2CB] =1/2[2CI+6CA+2CB]=C1+3CA+CB) القياس CA.103 CB.103 CNaCl K I 1 0.596 1.004 - 89 2.2x10-3 2 0.600 0.696 0.005 76 75x10-3 3 0.020 56 22.5x10-3 4 0.691 0.00 49 32.5x10-3
![I =1/2[C1x 12 +C1x12] +1/2[CAx22 +2CAX12] +1/2[CBX(1)2 +CBX12]](http://slideplayer.ae/slide/15551337/88/images/162/I+%3D1%2F2%5BC1x+12+%2BC1x12%5D+%2B1%2F2%5BCAx22+%2B2CAX12%5D+%2B1%2F2%5BCBX%281%292+%2BCBX12%5D.jpg "A + B → C + D. I =1/2[C1x 12 +C1x12] +1/2[CAx22 +2CAX12] +1/2[CBX(1)2 +CBX12] =1/2[2C1 ] + 1/2[4CA+2CA] +1/2[2CB] =1/2[2CI+6CA+2CB]=C1+3CA+CB) القياس. CA.103. CB.103. CNaCl. K. I x x x x10-3.")
163
2.إذا كانت قيمة ثابت سرعة التفاعل لتفاعل ما عند 30ºم هي ضعف قيمته عند 20ºم أحسبي طاقة التنشيط لهذا التفاعل. 3.أعطيت في أديبات إعتماد ثابت التفكك لفنيل أثيل إلى الأيثلين والاسيتالدهيد بالعلاقة: Kc=2.7x1011 exp(-43800/RT)Sec-1 أوجدي الانتروبي للتنشيط لهذا التفاعل . 4. أحســـبي ثابت الـــمعدل لتـفكك خامــس أكسيد النتروجين عند 65ºم. إذا كانت طاقة التنشيط لهذه العملية Eº=24.7 K.Cal قابل القيمة التي تحصل عليها بثابت المعدل الذي وجد عملياً. K=0.29min-1 علماً بأن ∆S= O ، c=تمثل التراكيز بالجرام أيون لتر-1.
Sec-1 أوجدي الانتروبي للتنشيط لهذا التفاعل . 4. أحســـبي ثابت الـــمعدل لتـفكك خامــس أكسيد النتروجين عند 65ºم. إذا كانت طاقة التنشيط لهذه العملية Eº=24.7 K.Cal قابل القيمة التي تحصل عليها بثابت المعدل الذي وجد عملياً. K=0.29min-1 علماً بأن ∆S= O ، c=تمثل التراكيز بالجرام أيون لتر-1.")
164
طاقة الحركة للتفاعلات الكيميائية
توجد عدة عوامل إلكترونية تستطيع أن تؤثر في نشاط مركب من المركبات في وضع معين أما الكيفية التي تؤثر بها هذه العوامل على التفاعلات واختلافها من مركب آخر فالمعلومات المتوفرة بهذا الخصوص قليلة وبالرغم من ذلك فهي تؤثر في نشاط التفاعل وفي طاقته الحركية وبذلك بتغير مجرى التفاعل وتختلف سرعته هذه الاعتبارات لها أهمية خاصة لأنها تستطيع أن تلقي الضوء على تفاصيل مجرى سير التفاعل. 1.الطاقة الحرة للتفاعل.. في أي تفاعل كيميائي يتم فيه تحويل المواد المتفاعلة إلى مواد ناتجة فأنه من الضروري معرفة المدى الذي يستمر فيه التفاعل ليعطي مواد ناتجة فالنظام عادة (يشمل مجموعة المواد المتفاعلة والنواتج معاً) يميل في اتجاه الحالة المستقرة، ولهذا يمكن التوقع أنه كلما كانت المواد الناتجة أقرب إلى الطور المستقر ( وذلك بموازنتها بالمواد المتفاعلة) استمر التفاعل في اتجاه إعطاء الناتج ولا يتوقع أن يحدث بين المواد المتفاعلة والمواد الناتجة أي توازن في هذه الحالة. ومعنى هذا أنه كلما كانت حالة الاستقرار للمواد الناتجة أكبر كما هي موضحة في الشكل التالي شكل (7) وضح وازداد التحويل إلى مواد ناتجة.
 يميل في اتجاه الحالة المستقرة، ولهذا يمكن التوقع أنه كلما كانت المواد الناتجة أقرب إلى الطور المستقر ( وذلك بموازنتها بالمواد المتفاعلة) استمر التفاعل في اتجاه إعطاء الناتج ولا يتوقع أن يحدث بين المواد المتفاعلة والمواد الناتجة أي توازن في هذه الحالة. ومعنى هذا أنه كلما كانت حالة الاستقرار للمواد الناتجة أكبر كما هي موضحة في الشكل التالي شكل (7) وضح وازداد التحويل إلى مواد ناتجة.")
165
والتغير البسيط في الطاقة الذي يحدث في عملية التحويل من المواد التي بدأ بها التفاعل إلى المواد التي نتجت عنه يمكن قياسه بسهولة باعتبار الفرق في الحرارة الكلية للتفاعل ΔH حيث H تمثل الحرارة الكلية الموجودة في مركب ما، أو هي ما يسمى بالمحتوى الحراري لمركب ما في وحدة كتلة (Enthalpy) ويسبق علامة ناقص إذا كان المحتوى الحرارية للمواد الناتجة أكثر انخفاضاً عما هو للمواد المتفاعلة، وعندما يوجد هذا النقص يطلق على التفاعل بأنه من النوع الطارد للحرارة، أما كان المحتوى الحراري للمواد الناتجة أعلى منه للمواد المتفاعلة فإنه يطلق على التفاعل بأنه من النوع الماص للحرارة. وتجدر الإشارة هنا إلى أن H ليست مقياساً مناسباً لفرق الاستقرار بين المواد المتفاعلة والمواد الناتجة، وذلك لأنه لا يوجد في الغالب ارتباط متبادل بين H وبين ثابت الاتزان للتفاعل K ولو أن أغلب التفاعلات الطاردة للحرارة بدرجة كبيرة لها ثوابت اتزان صغيرة وذلك لأن تحويل المواد المتفاعلة إلى مواد ناتجة قليل.

166
أما التفاعلات الماصة للحرارة فنجد أن ثوابت الاتزان لها كبيرة، ولزيادة الإيضاح، فإن هناك عوامل أخرى بالإضافة إلى عامل المحتوى الحرارية يجب أن يكون لها دور في الاستقرار النسبي ومنها ميل النظم المرتبة لتصبح نظم غير مرتبة ومعرفة درجة عدم الترتيب للنظام يمكن الحصول عليها من محددة التغيير الخاصة بالنظام والتي تسمى الانتروبي، ومن أجل قياس الاستقرار النسبي يتحتم علينا أن نأخذ أمراً وسطاً بين المحتوى الحراري (H) وبين الانتروبي (S) ويعبر عن العامل الوسط بالطاقة الحرة لجيبس حسب المعادلة: H = H - TS (92) حيث T هي درجة الحرارة المطلقة. التغير في الطاقة الحرة خلال التفاعل عند درجة الحرارة معينة يعبر عنه بالمعادلة التالية: ΔG = ΔH - TΔS (93) وقد وجد أن التغير في الطاقة الحرة عند تحويل المادة المتفاعلة لتعطي المادة الناتجة ΔGº يتناسب مع ثابت الاتزان K بالنسبة إلى التغير بحسب العلاقة: -ΔGº = RT log K (94) حيث ΔHº تدل على التغير تحت شروط قياسية أي لوزن جزئ واحد وعند ضغط واحد .
 وقد وجد أن التغير في الطاقة الحرة عند تحويل المادة المتفاعلة لتعطي المادة الناتجة ΔGº يتناسب مع ثابت الاتزان K بالنسبة إلى التغير بحسب العلاقة: -ΔGº = RT log K (94) حيث ΔHº تدل على التغير تحت شروط قياسية أي لوزن جزئ واحد وعند ضغط واحد .")
167
المعادلة (94) تدل على أن الزيادة في قيمة الطاقة الحرة السالبة (-ΔG) عند تحويل المواد المتفاعلة لتعطي المواد الناتجة تؤدي إلى زيادة قيمة (K) ويكون التفاعل بعيداً عن الاتزان ويتجه نحو التتابع لمصلحة المواد الناتجة. تتطلب الزيادة في قيمة الطاقة الحرة الموجبة ΔGº تناقضاً في القيمة الكسريه لثابت الاتزان (K) والعلاقة هنا من النوع اللوغاريتمي وتتفق مع تحول صغير للغاية في المواد المتفاعلة لتعطي مواد ناتجة والعكس صحيح. كما سبق أن ذكرنا بالنسبة إلى الزيادة في قيمة الطاقة الحرة ΔGº فإنها تتطلب بالمثل تزايداً كبيراً في قيمة ثابت الاتزان K وعلى هذا نجد أن قيمة المقدار الذي في حدود 10 كيلو سعر/وزن جزئ تقابل اتزان K=107 وهذا يتطلب بالضرورة إكمال تحول المواد المتفاعلة إلى مواد ناتجة. أي أنه بمعرفة الطاقة الحرة القياسية للمواد المتفاعلة والمواد الناتجة يمكن التنبؤ بالمدى المتوقع لتحول المواد المتفاعلة إلى مواد ناتجة. العامل (ΔH) من الممكن موازنته بالفرق في الطاقات للروابط بالنسبة للمواد المتفاعلة والمواد الناتجة والقيمة التقريبية لقيمة ΔH بالنسبة إلى أي تفاعل من المستطاع التنبؤ بها في أغلب الأحيان من جدول طاقات الروابط القياسية.
 والعلاقة هنا من النوع اللوغاريتمي وتتفق مع تحول صغير للغاية في المواد المتفاعلة لتعطي مواد ناتجة والعكس صحيح. كما سبق أن ذكرنا بالنسبة إلى الزيادة في قيمة الطاقة الحرة ΔGº فإنها تتطلب بالمثل تزايداً كبيراً في قيمة ثابت الاتزان K وعلى هذا نجد أن قيمة المقدار الذي في حدود 10 كيلو سعر/وزن جزئ تقابل اتزان K=107 وهذا يتطلب بالضرورة إكمال تحول المواد المتفاعلة إلى مواد ناتجة. أي أنه بمعرفة الطاقة الحرة القياسية للمواد المتفاعلة والمواد الناتجة يمكن التنبؤ بالمدى المتوقع لتحول المواد المتفاعلة إلى مواد ناتجة. العامل (ΔH) من الممكن موازنته بالفرق في الطاقات للروابط بالنسبة للمواد المتفاعلة والمواد الناتجة والقيمة التقريبية لقيمة ΔH بالنسبة إلى أي تفاعل من المستطاع التنبؤ بها في أغلب الأحيان من جدول طاقات الروابط القياسية.")
168
عامل النتروبي من غير المستطاع تفسيره بسهولة واضحة غير أنه يتعلق بفعالية عدد الطرق الممكنة التي يتم فيها إحداث طاقة كلية متجمعة يمكن تقسيمها بين تجمع من تجمعات الجزيئات وكذلك لعدد الطرق التي يتم بها توزيع طاقة الجزيئات الفردية بواسطة الحركات الانتقالية والدورانيه والاهتزازية ومن المحتمل أن تكون فيها الحركة الانتقالية أكبر في الكمية بشكل واضح، وعلى هذا لأي تفاعل من التفاعلات التي يكون فيها عدد الجزيئات التي تأخذ طريقها من الحالة المتفاعلة إلى الحالة الناتجة كبيراً كما في المعادلة الآتية: A B + C من المحتمل وجود زيادة كبيرة في الانتروبي (S) بسبب الكسب في حرية الحركة الانتقالية. المقدار –T ΔS من الممكن حينئذ أن يكون كبيراً لدرجة تسمح أن يشتمل على المقدار +ΔH للتفاعل الماص للحرارة وبذلك يصل إلى قيمة سالبة للطاقة الحرة –ΔG والاتزان يكون في جانب المواد الناتجة. إذا كان التفاعل طارد للحرارة فإن ΔH تكون سالبة، والطاقة الحرة ΔG أكثر سالبيه وثابت الاتزان K يتزايد بالمثل، أما إذا حدث نقص في عدد الجزيئات المشتركة في التفاعل عند تحولها من مواد متفاعلة إلى مواد ناتجة فمن المحتمل أن يحدث نقص في الأنتروبي –ΔS حسب المعادلة: ΔG = ΔH - (-) TΔS
 TΔS .")
169
ومالم يكن التفاعل طارداً للحرارة بدرجة كافية (أي أن ΔH سالبة وكبيرة بدرجة كافية) بحيث يحدث الاتزان فإن ΔH ستكون موجبة مما يؤدي لأن يكون الاتزان في جانب المواد المتفاعلة. التفاعلات الحلقية يمكن أن تتم بنقص في الأنتروبي (رغماً عن كون هذا التفاعل ينقص فيه عدد الجزيئات المشتركة في التفاعل عند تكوين المواد الناتجة) كما في المثال الآتي: وبالرغم من عدم وجود فرق كبير في أنتروبي الحركة الانتقالية فإنه يجب إدخال ثابت يقابل التغيير بسبب الحركة الدورانيه حول الروابط بين ذرات الكربون التي تكون منطلقة في السلاسل المفتوحة للمادة المتفاعلة إلا أنها مقيدة بدرجة كبيرة في المركبات الحلقية الناتجة. من الواجب عدم التغاضي عن أن مصطلح انتروبي يتضمن درجة الحرارة TΔS بينما مصطلح المحتوى الحراري ΔH لا يشمل درجة الحرارة كما أن إسهاماتها النسبية لتغير الطاقة الحرة تختلف اختلافاً ملموساً بالنسبة إلى التفاعل الواحد إذا أخذ مكانه عند درجات حرارة مختلفة على نطاق واسع.
 كما في المثال الآتي: وبالرغم من عدم وجود فرق كبير في أنتروبي الحركة الانتقالية فإنه يجب إدخال ثابت يقابل التغيير بسبب الحركة الدورانيه حول الروابط بين ذرات الكربون التي تكون منطلقة في السلاسل المفتوحة للمادة المتفاعلة إلا أنها مقيدة بدرجة كبيرة في المركبات الحلقية الناتجة. من الواجب عدم التغاضي عن أن مصطلح انتروبي يتضمن درجة الحرارة TΔS بينما مصطلح المحتوى الحراري ΔH لا يشمل درجة الحرارة كما أن إسهاماتها النسبية لتغير الطاقة الحرة تختلف اختلافاً ملموساً بالنسبة إلى التفاعل الواحد إذا أخذ مكانه عند درجات حرارة مختلفة على نطاق واسع.")
170
.تنشيط التفاعل: بالرغم من أن سالبية الطاقة الحرة ΔGº شرط لازم عند حدوث تغيير بشكل ما تحت شروط معينة فإن ظهور الطاقة الحرة في حد ذاتها غير كافية لأن تخبرنا عن الشرعة التي تتحول بها المواد المتفاعلة إلى مواد ناتجة أي أنه رغماً عن كونها تنبأ بالمدى المتوقع لتحول المواد المتفاعلة إلى مواد ناتجة إلا أنها لا تذكر شيئاً عن سرعة التفاعل وهل هو سريع أو تفاعل بطئ فمثلاً عند أكسدة السليلوز حسب التفاعل الآتي: (C6H10O5)n + 6(n)O2 6(n)CO2 + 5(n)H2O نجد أن ΔGº لهذا التفاعل سالبيه وكبيرة في قيمتها لدرجة أن الاتزان يقع بالضرورة وقوعاً تاماً في جانب تكون غاز ثاني أكسيد الكربون والماء (المواد الناتجة) ولكن إذا أخذنا أي صحيفة من الصحف (تتكون من السليلوز بدرجة كبيرة) من الممكن قراءتها في جو من الهواء (أو حتى في جو من الأكسجين) في درجة حرارة الغرفة وذلك لفترات زمنية طويلة دون أن يلاحظ عليها أي تغيير أو تحول إلى منتجات غازية، نستنتج من هذا أن سرعة التفاعل بطيئة للغاية تحت هذه الظروف بصرف النظر عن كبر قيمة ΔHº وهذا التفاعل إذا تغيرت الظروف والشروط له نجده سريعاً جداً عند درجات الحرارة العالية.
n + 6(n)O2 6(n)CO2 + 5(n)H2O. نجد أن ΔGº لهذا التفاعل سالبيه وكبيرة في قيمتها لدرجة أن الاتزان يقع بالضرورة وقوعاً تاماً في جانب تكون غاز ثاني أكسيد الكربون والماء (المواد الناتجة) ولكن إذا أخذنا أي صحيفة من الصحف (تتكون من السليلوز بدرجة كبيرة) من الممكن قراءتها في جو من الهواء (أو حتى في جو من الأكسجين) في درجة حرارة الغرفة وذلك لفترات زمنية طويلة دون أن يلاحظ عليها أي تغيير أو تحول إلى منتجات غازية، نستنتج من هذا أن سرعة التفاعل بطيئة للغاية تحت هذه الظروف بصرف النظر عن كبر قيمة ΔHº وهذا التفاعل إذا تغيرت الظروف والشروط له نجده سريعاً جداً عند درجات الحرارة العالية.")
171
عملية تحول المواد المتفاعلة إلى مواد ناتجة بغض النظر عن سالبيه ΔGº من المتعذر أن تحدث وكأنها سريان من مرتفع إلى أسفل كما هو موضح في الشكل (8- أ) وإنما هناك حاجز للطاقة يعترض طريقها وعليها أن تتغلب عليه كما هو في الشكل (8- ب). 3. معدل التفاعل والطاقة الحرة للتنشيط.. الموقع (x) في الشكل (رقم 8– ب) للطاقة الحرة يمثل موقع التشكل للطور الأقل ثباتاً الذي تمر به المواد المتفاعلة في طرق تحولها إلى مواد ناتجة وعموماً هذا التشكل يسمى بالمركب النشط أو حالة التحول الانتقالية. ويجب التأكيد على أن هذا ليس إلا مجرد حالة عدم استقرار بدرجة عالية والتي تمر بها العملية الديناميكية وليس حالة متوسطة بمعنى أنه لا يمكن فعلاً فصلها أو عزلها ولكن فقط حالة عدم استقرار تتوسط بين المواد المتفاعلة والمواد الناتجة ويمكن توضيح ذلك بالمثال الآتي:
 في الشكل (رقم 8– ب) للطاقة الحرة يمثل موقع التشكل للطور الأقل ثباتاً الذي تمر به المواد المتفاعلة في طرق تحولها إلى مواد ناتجة وعموماً هذا التشكل يسمى بالمركب النشط أو حالة التحول الانتقالية. ويجب التأكيد على أن هذا ليس إلا مجرد حالة عدم استقرار بدرجة عالية والتي تمر بها العملية الديناميكية وليس حالة متوسطة بمعنى أنه لا يمكن فعلاً فصلها أو عزلها ولكن فقط حالة عدم استقرار تتوسط بين المواد المتفاعلة والمواد الناتجة ويمكن توضيح ذلك بالمثال الآتي: .")
172
في التحليل المائي القلوي لبروميد الميثيل CH3Br الذي تتكون فيه رابطة C-C--OH) قبل أن تنقطع رابطة (Br..C) تماماً وكذلك ذرات الهيدروجين الثلاث المتصلة بذرة الكربون التي تمر خلال التشكل نجدها جميعاً تقع في مستوى واحد. في المثال السابق المركب النشط المتوسط يمثل حالة التحول الانتقالية وهي تمثل الطور غير الثابت الذي لا يمكن فصله أو عزله. كذلك نجد في الشكل أن ارتفاع حاجز الطاقة ΔG≠ يمثل الطاقة الحرة لتنشيط التفاعل وكلما كان الحاجز مرتفعاً كان التفاعل بطيئاً ويتكون هذا الحاجز من المحتوى ΔH≠ ومن الانتروبي ΔS≠ كالتالي: ΔG≠ = ΔH≠ - TΔS≠ المحتوى الحراي للتنشيط ΔH≠ يطابق الطاقة اللازمة لإمتطاط أو حتى تكسير الروابط التي هي من المستلزمات الأولية ليأخذ التفاعل مجراه كما في المثال السابق عند إمتطاط الرابطة بين ذرة الكربون وذرة البروم (C-Br) . لذا يتحتم على الجزيئات الداخلة في التفاعل أن تتصادم بدرجة يتكون نتيجتها حد أدنى من الطاقة يمكن التفاعل من أن يأخذ مكانه ويستمر وتسمى هذه بطاقة التنشيط وتعطي أحياناً الرمز ΔE ولكنها متساوية مع ΔH≠ عند إرتفاع درجة الحرارة للتفاعل نلاحظ زيادة واضحة في معدل سير التفاعل نتيجة للنسبة المتزايدة من الجزيئات التي لها طاقة أعلى من الحد الأدنى بقدر ارتفاع درجة الحرارة.
 . لذا يتحتم على الجزيئات الداخلة في التفاعل أن تتصادم بدرجة يتكون نتيجتها حد أدنى من الطاقة يمكن التفاعل من أن يأخذ مكانه ويستمر وتسمى هذه بطاقة التنشيط وتعطي أحياناً الرمز ΔE ولكنها متساوية مع ΔH≠ عند إرتفاع درجة الحرارة للتفاعل نلاحظ زيادة واضحة في معدل سير التفاعل نتيجة للنسبة المتزايدة من الجزيئات التي لها طاقة أعلى من الحد الأدنى بقدر ارتفاع درجة الحرارة.")
173
4.الطاقة الحركية والخطوة المحددة لسرعة التفاعل: يمكن قياس معدلات التفاعل بفحص معدل اختفاء المواد المتفاعلة أو معدل ظهور المواد الناتجة عند درجة حرارة معينه (ثابتة) وذلك سعياً وراء أن ينسب هذا إلى تركيز أحد المواد المشتركة في التفاعل أو جميعها. ويمكن تتبع التفاعل بعدة طرق مثل المعايرة بالتحليل الحجمي والقياس الطيفي والقياس اللوني وغير ذلك.. ولكن الخطوة القاطعة تتضمن اختبار معلومات أو بيانات الطاقة الحركية الخام عند مختلف مراحل التركيز الممكنة حتى يصل الأمر في النهاية إلى الحصول على نتيجة مقبولة ومعقولة فالتفاعل الآتي.. CH3Br + OH- CH3OH+Br هذا التفاعل من الدرجة الثانية بالنسبة إلى التفاعل الكلية ومن الدرجة الأولى بالنسبة إلى كل من بروميد وأيون الهيروكسيد، حيث K هي ثابت سرعة التفاعل. R= K [CH3 – Br] [OH-] في أغلب الأحيان نجد أن معادلات تحديد إتحاد المواد الكيميائية ليست مرشداً مناسباً للتعرف على المواد المؤثرة في سرعة التفاعل فمثلاً في تفاعل برومه الأسيتون في الوسط القلوي كما في المعادلة: OH- CH3 – C – CH3 + Br2 CH3COCH2Br + HBr O تكون معادلة سرعة التفاعل السابق : R =K[CH3COCH3][OH-] أي أن البروم في الظاهر لا يؤثر في سرعة التفاعل هو قلوية المحلول، ومن الواضح أن البروم لابد وأن يدخل في بعض مراحل التفاعل الإجمالي حيث أن هذه المادة تكون مندمجة في الناتج النهائي CH3COCH2Br ولكنها غير داخلة في المرحلة التي تقاس فعلاً سرعة التفاعل.

174
مما سبق نجد أن معادلات تحديد اتجاه المواد الكيميائية لأي تفاعل ليس من شأنها أن تعطي أي دليل مهما كان بخصوص خط السير الواقعي الذي يسلكه التفاعل ويمكن توضيح ذلك بالمثال الآتي: 6CH2O + 4NH3 C6H12N4 + 6H2O في التفاعل السابق الخاص بتكوين الهكسامين نجد أن فرصة التصادم الآتي في الوقت نفسه لسته جزيئات من CH2O وأربعة جزيئات من الأمونيا لا يمكن أن تحدث لأن صدمة قوامها عشرة أجسام لا وجود لها من الناحية الفعلية. ولكنه حتى عندما يكون تحديد إتحاد المواد الكيميائية أقل تطرفاً فإن التفاعلات عادة تتألف من عدد من الخطوات المتتابعة ومن هذه الخطوات خطوة بطيئة يجري في الواقع قياسها وبذلك تعمل على تحديد سرعة التفاعل وبذلك تسمى الخطوة المحددة لسرعة التفاعل. في الشكل (9) نجد أن المواد الأولية (المتفاعلة) حول عن طريق الانتقال (X2)إلى حلة متوسطة والتي تتفكك بعد ذلك إلى مواد ناتجة عن طريق حالة الانتقال (X2). من الشكل نجد أن تكوين الحالة المتوسطة عن طريق (X1) يتطلب طاقة أكثر ∆G2# < ∆G1# من الخطوة الثانية وبذلك تكون الخطوة الأولى هي الخطة البطيئة والتي تتحكم في سير التفاعل أي أنها المرحلة التي تقاس سرعتها عملياً في تجارب الكيمياء الحركية. وتتبعها خطوة سريعة أقل احتياجاً للطاقة وهي تحويل الحالة المتوسطة إلى ماد ناتجة وهذه الخطوة الأخيرة غير محددة لسرعة التفاعل.
 نجد أن المواد الأولية (المتفاعلة) حول عن طريق الانتقال (X2)إلى حلة متوسطة والتي تتفكك بعد ذلك إلى مواد ناتجة عن طريق حالة الانتقال (X2). من الشكل نجد أن تكوين الحالة المتوسطة عن طريق (X1) يتطلب طاقة أكثر ∆G2# < ∆G1# من الخطوة الثانية وبذلك تكون الخطوة الأولى هي الخطة البطيئة والتي تتحكم في سير التفاعل أي أنها المرحلة التي تقاس سرعتها عملياً في تجارب الكيمياء الحركية. وتتبعها خطوة سريعة أقل احتياجاً للطاقة وهي تحويل الحالة المتوسطة إلى ماد ناتجة وهذه الخطوة الأخيرة غير محددة لسرعة التفاعل.")
175
من الممكن تحت شروط معينه أن يقال عن تفاعل برومه الاسيتون الموضح سابقاً أنه يتبع نمطاً مثالياً مطابقاً للشكل الذي يتم فيه إزالة البروتون وهي خطوة بطيئة ومحددة لسرعة التفاعل وذلك لتكوين الحالة المتوسطة التي تتلوها خطوة سريعة وفيها يداهم البروم المركب ليعطي المواد الناتجة وهي برومو اسيتون وأيون البروميد وذلك بحسب المعادلة التالية: OH- + H-CH2COCH3 Slow step H2O + CH2COCH3 Fast step Br- + Br – CH – COOH3 ولابد من الإشارة أنه بالرغم من أن التفسير السابق يتسم بالاستنتاج المعقول نتيجة التجارب العملية التي تقوم عليها معادلة سرعة التفاعل فإن الأخيرة لا يمكن أن يقال أنها تبرهن الأولى. كما أن تأثير الحوافز في زيادة السرعة التي يسير بها التفاعل تكمن في تزويد التفاعل بمسار بديل له طاقة تنشيطية منخفضة وغالباً ما يحدث هذا خلال تكوين متوسط جديد يكون أكثر استقرارً وله طاقة منخفضة (شكل 10).
.")
176
ومن الأمثلة على ماسبق نجد أن معدل التحلل المائي للألكين مباشرة بالماء غالباً بطئ بدرجة واضحة ومعادلة التفاعل كالتالي: ولكن من الممكن إسراع التفاعل بدرجة كبيرة وذلك بإضافة حمض فيجري التفاعل في وسط حمضي ويقوم الحمض بدور العامل المساعد الذي له التأثير البروتوني الأولي في الألكين فيحوله إلى أيون كاربوني متوسط ويتلو هذا مداهمة سريعة وبسيطة على الأيون المشحون بشحنة موجبة من جزئ الماء حيث يتحرر البروتون ليؤدي وظيفة العامل المساعدة مرة ثانية كما في المعادلات التالية:

177
.إستطلاع ميكانيكية التفاعل: يندر إن لم يكن من المتعذر إعداد بيانات كاملة وصحيحة فيما يتصل بالنواحي التركيبية والتنشيطية والكيمياء المجسمة عن المسار الذي يسلكه أي تفاعل كيميائي كما أنه أمر صعب، ومع ذلك فمن المستطاع جمع بيانات ومعلومات كافية تثبيت بها أن أحد أنواع الميكانيزم الممكن نظرياً لا يلائم بالضبط نتائج التجارب العملية أو تثبيت بها أنها تعمل على إظهار أفضلية أحد الاختيارات المتعددة وتقف في صفة دون غيره، وعليه يمكن دراسة المؤثرات الآتية: أ.طاقة الحركة: إن أغلب المعلومات التي يمكن الحصول عليها مصدرة دراسة طاقة الحركة 1/2mR2 غير أن تفسير بيانات ومعلومات طاقة الحركة باستخدام مصطلحات الميكانيكية ليست دائماً سهلة أو تتفق مع ما تم فرضه، وعلى ذلك فالجزيئات الداخلة في التفاعل ولها فعاليتها فيه والتي تركيزها يحدد سرعة التفاعل قد تختلف تماماً عن خليط التفاعل الذي بدأ به التفاعل، ففي النترجة للمركبات العطرية المجموعة المداهمة هي عادة NO2+ ولكن المركب الذي استخدم في مخلوط التفاعل هو حمض النيتريك وهو الذي يقاس التغيير في تركيزه، العلاقة بين الاثنين قد تكون علاقة تتسم بالتعقيد ولهذا السبب فإن استنتاج سرعة التفاعل من قياس التغيير في تركيز حمض النيتريك مسألة تحتاج إلى مراجعة، أي أنه بصرف النظر عن حقيقة أن هذا التفاعل هو تفاعل بسيط فإنه قد لا يكون من السهولة استنتاج سرعة التفاعل من المقادير التي يمكن قياسها مباشرة.

178
كذلك التحليل المائي للهاليدات الألكيل Rx وجد أنها تتبع معادلة سرعة التفاعل الآتية: R = K[R-X] وعليه فليس من الضروري الوصول إلى أن خطوة تحديد سرعة التفاعل لا تشمل اشتراك الماء على أساس بسيط هو أن الماء لم يظهر في معادلة سرعة التفاعل ذلك لأنه في حالة استخدام كمذيب فإن تركيزه سيبقى فعلاً بدون تغيير يذكر سواء اشترك أو لم يشترك في الخطوة المحددة للسرعة. ويمكن توضيح دور الماء فيمكن إجراء التفاعل باستخدام مذيب آخر مثلاً حمض الفورميك (H-COOH) ، ويضاف الماء بتركيز قليل جداً كنواة فيلية (Necleo Phile) وعندها نجد أن معادلة سرعة التفاعل تكون كالتالي: R = K[R-X] [H2O] غير أن الميكانيكية الواقعية لتفاعل التحلل من الممكن أن تتغير بتغير المذيب ولهذا فليس من الضروري التركيز على كيفية ما حدث في المحلول المائي. الغالبية العظمى للتفاعلات العضوية يتم إجراؤها في محاليل ولذلك نجد أن التغيرات البسيطة جداً في المذيب يكون لها تأثير عميق في سرعة التفاعل وميكانيكيته ولاسيما المركبات المتوسطة القطبية مثل مكونات أزواج الأيون هذه المركبات عادة تحمل معها غمدها من جزيئات المذيب والذي من شأنه أن يؤثر كثيراً في استقرارها وبهذا نجد أن التفاعل يتأثر بقوة كبيرة بتركيب المذيب المستخدم وطبيعته ولاسيما من ناحية قطبية والقدرات المتصلة بالتذاوب الأيوني.
![كذلك التحليل المائي للهاليدات الألكيل Rx وجد أنها تتبع معادلة سرعة التفاعل الآتية: R = K[R-X] وعليه فليس من الضروري الوصول إلى أن خطوة تحديد سرعة التفاعل لا تشمل اشتراك الماء على أساس بسيط هو أن الماء لم يظهر في معادلة سرعة التفاعل ذلك لأنه في حالة استخدام كمذيب فإن تركيزه سيبقى فعلاً بدون تغيير يذكر سواء اشترك أو لم يشترك في الخطوة المحددة للسرعة.](http://slideplayer.ae/slide/15551337/88/images/178/%D9%83%D8%B0%D9%84%D9%83+%D8%A7%D9%84%D8%AA%D8%AD%D9%84%D9%8A%D9%84+%D8%A7%D9%84%D9%85%D8%A7%D8%A6%D9%8A+%D9%84%D9%84%D9%87%D8%A7%D9%84%D9%8A%D8%AF%D8%A7%D8%AA+%D8%A7%D9%84%D8%A3%D9%84%D9%83%D9%8A%D9%84+Rx+%D9%88%D8%AC%D8%AF+%D8%A3%D9%86%D9%87%D8%A7+%D8%AA%D8%AA%D8%A8%D8%B9+%D9%85%D8%B9%D8%A7%D8%AF%D9%84%D8%A9+%D8%B3%D8%B1%D8%B9%D8%A9+%D8%A7%D9%84%D8%AA%D9%81%D8%A7%D8%B9%D9%84+%D8%A7%D9%84%D8%A2%D8%AA%D9%8A%D8%A9%3A+R+%3D+K%5BR-X%5D+%D9%88%D8%B9%D9%84%D9%8A%D9%87+%D9%81%D9%84%D9%8A%D8%B3+%D9%85%D9%86+%D8%A7%D9%84%D8%B6%D8%B1%D9%88%D8%B1%D9%8A+%D8%A7%D9%84%D9%88%D8%B5%D9%88%D9%84+%D8%A5%D9%84%D9%89+%D8%A3%D9%86+%D8%AE%D8%B7%D9%88%D8%A9+%D8%AA%D8%AD%D8%AF%D9%8A%D8%AF+%D8%B3%D8%B1%D8%B9%D8%A9+%D8%A7%D9%84%D8%AA%D9%81%D8%A7%D8%B9%D9%84+%D9%84%D8%A7+%D8%AA%D8%B4%D9%85%D9%84+%D8%A7%D8%B4%D8%AA%D8%B1%D8%A7%D9%83+%D8%A7%D9%84%D9%85%D8%A7%D8%A1+%D8%B9%D9%84%D9%89+%D8%A3%D8%B3%D8%A7%D8%B3+%D8%A8%D8%B3%D9%8A%D8%B7+%D9%87%D9%88+%D8%A3%D9%86+%D8%A7%D9%84%D9%85%D8%A7%D8%A1+%D9%84%D9%85+%D9%8A%D8%B8%D9%87%D8%B1+%D9%81%D9%8A+%D9%85%D8%B9%D8%A7%D8%AF%D9%84%D8%A9+%D8%B3%D8%B1%D8%B9%D8%A9+%D8%A7%D9%84%D8%AA%D9%81%D8%A7%D8%B9%D9%84+%D8%B0%D9%84%D9%83+%D9%84%D8%A3%D9%86%D9%87+%D9%81%D9%8A+%D8%AD%D8%A7%D9%84%D8%A9+%D8%A7%D8%B3%D8%AA%D8%AE%D8%AF%D8%A7%D9%85+%D9%83%D9%85%D8%B0%D9%8A%D8%A8+%D9%81%D8%A5%D9%86+%D8%AA%D8%B1%D9%83%D9%8A%D8%B2%D9%87+%D8%B3%D9%8A%D8%A8%D9%82%D9%89+%D9%81%D8%B9%D9%84%D8%A7%D9%8B+%D8%A8%D8%AF%D9%88%D9%86+%D8%AA%D8%BA%D9%8A%D9%8A%D8%B1+%D9%8A%D8%B0%D9%83%D8%B1+%D8%B3%D9%88%D8%A7%D8%A1+%D8%A7%D8%B4%D8%AA%D8%B1%D9%83+%D8%A3%D9%88+%D9%84%D9%85+%D9%8A%D8%B4%D8%AA%D8%B1%D9%83+%D9%81%D9%8A+%D8%A7%D9%84%D8%AE%D8%B7%D9%88%D8.jpg "ويمكن توضيح دور الماء فيمكن إجراء التفاعل باستخدام مذيب آخر مثلاً حمض الفورميك (H-COOH) ، ويضاف الماء بتركيز قليل جداً كنواة فيلية (Necleo Phile) وعندها نجد أن معادلة سرعة التفاعل تكون كالتالي: R = K[R-X] [H2O] غير أن الميكانيكية الواقعية لتفاعل التحلل من الممكن أن تتغير بتغير المذيب ولهذا فليس من الضروري التركيز على كيفية ما حدث في المحلول المائي. الغالبية العظمى للتفاعلات العضوية يتم إجراؤها في محاليل ولذلك نجد أن التغيرات البسيطة جداً في المذيب يكون لها تأثير عميق في سرعة التفاعل وميكانيكيته ولاسيما المركبات المتوسطة القطبية مثل مكونات أزواج الأيون هذه المركبات عادة تحمل معها غمدها من جزيئات المذيب والذي من شأنه أن يؤثر كثيراً في استقرارها وبهذا نجد أن التفاعل يتأثر بقوة كبيرة بتركيب المذيب المستخدم وطبيعته ولاسيما من ناحية قطبية والقدرات المتصلة بالتذاوب الأيوني.")
179
أما التفاعلات التي تضم مجموعات جذور فهي أقل تأثراً بطبيعة المذيب مالم تكن قادرة على التفاعل مع الجذور هذه التفاعلات تتأثر بدرجة كبيرة بإضافة المصادر للجذور (مثلاً مركبات فوق الأكاسيد) أو بإضافة ممتصات المجموعات الجذرية مثل الكينونز أو بإجراء التفاعل في وجود الضوء الذي يمكن أن يساعد على بدء التفاعل عن طريق إنتاج المجموعات الجذرية بواسطة التنشيط الكيميائي الضوئي (Photo – Chemical). ب. تأثير المتماكنات: حيث أن بيانات طاقات الحركة البسيطة لا تعطي أي فكرة حول رابطة معينه أكسرت في مرحلة تحديد سرعة التفاعل أو لا، ولذلك يتحتم الالتجاء إلى تحميص أكثر على سبيل المثال إذا كانت الرابطة المعينة هي (C-H) فإنه بموازنة سرعات التفاعل تحت الشروط نفسها بين المركب تحت الدراسة وبين المركب المماثل المحتوى على نظير أثقل مثلاً ديوتيروم (C-D) الذي فيه رابطة أصعب من أن تكسر ولذلك يكون أبطأ تقريباً بمقدار 7 مرات عند 25ºم وعلى هذا ففي عملية الأكسدة التالية:
 فإنه بموازنة سرعات التفاعل تحت الشروط نفسها بين المركب تحت الدراسة وبين المركب المماثل المحتوى على نظير أثقل مثلاً ديوتيروم (C-D) الذي فيه رابطة أصعب من أن تكسر ولذلك يكون أبطأ تقريباً بمقدار 7 مرات عند 25ºم وعلى هذا ففي عملية الأكسدة التالية:")
180
Ph2CHOH Ph2C=O + H2I نجد أن هذا التفاعل يتأثر بالبرمنجنيات القلوية، كما وجد أيضاً أن (Ph2CHOH) تتأكسد بمقدار 6.7 مرة أسرع من (Ph2CDOH) في هذا المثال نجد بوضوح أن كسر رابطة (C-H) قد اشتملت عليها خطوة تحديد سرعة التفاعل، ولذلك يقال عن هذا التفاعل أنه يعرض بوضوح تأثير المتماكنات. خلافاً لهذا المثال نجد أن البنزين (C6H6) والهكساديوتوبنزين (C6D6) عند معالجتها بمجموعة (NO2+) يسيران بالمعدل نفسه مما يدل على أنه كسر رابطة (C- H) قد أخذت مكانها في إحدى مراحل التفاعل الكلية ولكن لم تشتمل عليها خطوة تحديد سرعة التفاعل، ولذلك لم يظهر لها تأثير في السرعة.
 والهكساديوتوبنزين (C6D6) عند معالجتها بمجموعة (NO2+) يسيران بالمعدل نفسه مما يدل على أنه كسر رابطة (C- H) قد أخذت مكانها في إحدى مراحل التفاعل الكلية ولكن لم تشتمل عليها خطوة تحديد سرعة التفاعل، ولذلك لم يظهر لها تأثير في السرعة.")
181
المتماكنات يمكن استخدامها لحل مسائل تتعلق بميكانيكية التفاعل وحدها دون طاقة الحركة، وعلى ذلك فالتحلل المائي للأسترات لتعطي حمضاً وكحولاً يمكن أن تسير من الوجهة النظرية بالانشطار عن طريق (1) وهو إنشطار إلكيل-أوكسجين أو بالإنشطار عن طريق (2) وهو إنشطار أسايل – أوكسجين كما يلي: الماء الثقيل (D2O) قد غلب استعماله في طريقة مشابهة إلى حد ما كما يرى في تفاعل كانيزارو للبنزالدهيد التالي: لمعرفة هل ذرة الهيدروجين الثانية المتصلة بذرة الكربون والتي تنجذب إليها في جزئ كحول البنزايل المتكون آتية من المذيب H2O أو من جزئ آخر من البنزالدهيد، فقد أجرى التفاعل باستخدام الماء الثقيل D2O ووجد أنه لايؤدي إلى تكوين PhCHDOH مما يفسر ويوضح أن ذرة الهيدروجين الثانية لا يمكن أن يكون مصدرها الماء بل يتحتم أن يكون مصدرها جزئ ثان من البنزالدهيد.
 قد غلب استعماله في طريقة مشابهة إلى حد ما كما يرى في تفاعل كانيزارو للبنزالدهيد التالي: لمعرفة هل ذرة الهيدروجين الثانية المتصلة بذرة الكربون والتي تنجذب إليها في جزئ كحول البنزايل المتكون آتية من المذيب H2O أو من جزئ آخر من البنزالدهيد، فقد أجرى التفاعل باستخدام الماء الثقيل D2O ووجد أنه لايؤدي إلى تكوين PhCHDOH مما يفسر ويوضح أن ذرة الهيدروجين الثانية لا يمكن أن يكون مصدرها الماء بل يتحتم أن يكون مصدرها جزئ ثان من البنزالدهيد.")
182
ج.المتوسطات: إن الفرز الحقيقي لخليط التفاعل من عينة متوسطة أو أكثر في تفاعل تحويل المواد التي بدأ بها التفاعل إلى نواتج عطي براهين تتسم بالتماسك والثبات يمكن الاعتماد عليها لدراسة ميكانيكية التفاعل، ولو أن هذا الفرز يتعذر تحقيقه دائماً لسرعة تحول المركبات المتوسطة. O OH- R – C – NH2 RNH2 ومن أمثلة هذا النوع من الدراسة نجد في تفاعل هوفمان الذي تكون نتيجته تحويل الأميدات إلى أمينات كما يلي: OH- RNH2 R – C – NH2 يمكن مع العناية والحذر الفصل N بروموأمايد (R-CONHBR) وأيونه السالب (RCONBr) كذلك أيزووسيانات المركب (RNCO) وبهذا تتكون ميكانيكية هذا التفاعل قد اتضحت إلى حد كبير ويمكن افتراضها كالتالي:
 وأيونه السالب (RCONBr) كذلك أيزووسيانات المركب (RNCO) وبهذا تتكون ميكانيكية هذا التفاعل قد اتضحت إلى حد كبير ويمكن افتراضها كالتالي: .")
183
ويجب عموماً التأكد من أن أية عينات يعمل على فرزها فرزاً واقعياً هي من نوع المتوسطات وليست مجرد نواتج بديلة في التفاعل وذلك باختبار إمكان تحويلها إلى نواتج التفاعل المعتادة بسرعة تعادل على الأقل سرعة التفاعل إجمالياً تحت شروط التفاعل العادية نفسها ..كما أن عدم التمكن من فصل أي متوسط على الإطلاق لا يعني بالضرورة عدم تكوين شي من هذه المتوسطات بل أنها فقط متغيرة وغير ثابتة وقابلة للتحويل بسرعة مما لايساعد على فرزها ويستدل على وجود هذه المتوسطات غالباً بواسطة قياسات فيزيائية وبصفة خاصة القياسات الطبيعية ففي تكوين الأوكسيمات من عدد من المركبات الكربونية بالتفاعل مع الهيدروكسيل أمين كما يلي:

184
إن خصائص امتصاص حزمة الأشعة تحت الحمراء للمجموعة C=O في المادة التي يبدأ بها التفاعل تختفي بسرعة، وقد يكون الاختفاء كلية قبل ظهور الخصائص المميزة لامتصاص حزمة الأشعة بالمجموعة C=N- في ناتج التفاعل. ومن الواضح ضرورة تكوين أحد المتوسطات وكدليل إضافي تفسيري يقترح تكون المركب كالتالي: يتم تكوينه بسرعة، ثم يتفكك ببطء ليعطي النواتج التي تتمثل في الأوكسيم والماء، أما إذا توفر سبب وجيه للشك أو الارتياب في وجود عينه معينة بالذات كمركب متوسط غير ثابت وقابل للتحويل في سير تفاعل من التفاعلات فقد يكون من الممكن التأكد إزالة هذا الشك إدخال عينة متفاعلة مع خليط التفاعل وهذه العينة يجب أن نتوقع سلفاً أن المتوسط الذي سبق إفتراض وجوده سيتفاعل معها بسهولة ويسر. وهذا من قبيل التحاليل المدبر من أجل تعيين المركب المتوسط وفرزه، وعلى هذا ففي التحليل المائي لمادة الكلوروفورم بفعل القلويات القوية اقترح أن تكون مادة داي كلوروكاربين (Cl-C-Cl) ذات القصور الالكتروني العالي والتي اقترحت أن تكون مادة متوسطة غير ثابتة.
 ذات القصور الالكتروني العالي والتي اقترحت أن تكون مادة متوسطة غير ثابتة.")
185
للتأكد من هذا الاقتراح وإزالة الشك يمكن إدخال مادة غنية بالالكترونات في خليط التفاعل مثل (cis-but 2 ene) كما في الشكل (11-أ) ثم يتبع ذلك فرز المكونات الثابتة الناتجة لمشتقات السيكلوبروبان كما في الشكل (11-ب) والتي يتعذر تكوينها بأي طريقة أخرى مما يدل على وجود المركب المتوسط (CCl2) .
 كما في الشكل (11-أ) ثم يتبع ذلك فرز المكونات الثابتة الناتجة لمشتقات السيكلوبروبان كما في الشكل (11-ب) والتي يتعذر تكوينها بأي طريقة أخرى مما يدل على وجود المركب المتوسط (CCl2) .")
186
د.المعايير الكيميائية المجسمة : المعلومات عن التفاعلات الكيميائية المجسمة التي يسير عليها تفاعل معين بالذات من الممكن أن تعطي فكرة واضحة إلى حد ما عن الميكانيكية التي تم بها التفاعل كما قد تتسبب في إدخال معايير إضطرارية لتفسير ما يحدث في تفاعل ما، فالحقيقة الخاصة بدور الوساطة القلوية لبرومة الكيتون النشط ضوئياً، كما في الشكل التالي: ± ± PhCOCHMeEt PhCOC BrMeEt تؤدي إلى تكوين ناتج خامل ضوئياً مما يبين أن التفاعل من المحتم سيره خلال مادة متوسطة مستوية والتي يمكن أن تداهم بالتساوي من الطرفين لتنتج مقدارين متكافئيين من شكل صورتي المرآة للمادة الناتجة. كما أن السيكوبنتين يضيف البروم تحت شروط قطبية ليعطي ترانس ثنائي بروميد مما يوضح أن ميكانيكية التفاعل ليست مجرد إضافة بسيطة مباشرة لجزئ البروم إلى الرابطة المزدوجة إذ يتحتم معه الحصول على سيس ثنائي بروميد كما يجب أن تتم العملية في مرحلتين على الأقل هي: Br 𝑂𝐻

187
وهذا يوضح الحدود التي تتمشى معها ميكانيكية التفاعل.
كذلك نجد أن العديد من التفاعلات الحذف تأخذ مكانها مع الجزء العابر من زوج المتشابهات السيس والترانس كالذي يحدث عند تحويل مركبات خلات الالدوكسيم إلى مركب النترايل المعروف كما يلي: وهذا يوضح الحدود التي تتمشى معها ميكانيكية التفاعل.

188
هـ. اختلاف النتائج عن المتوقع: المعلومات الأساسية التي يحصل عليها عن أي تفاعل من تركيب الناتج أو النواتج فعلاً ثم إيجاد العلاقة التي تربط تركيبها بتركيب المواد المتفاعلة، قد يكون لهذه المعلومات دور إجباري وبصفة خاصة في حالة الحصول على ناتج غير متوقع على الإطلاق فمثلاً في تفاعل بارا-كلورتلوين مع أيون الامايد (NH2) في محلول النشادر لا يتم الحصول على بارا تولودين المتوقع وجوده فقط بل نجد أيضاً المركب غير المتوقع الحصول عليه إطلاقاً وهو ميتاً تولودين أمين من بارا – كلوروتلوين بطريقة الإحلال البسيطة غير ممكن، ويكون تكوينه اما عن طريق يختلف تماماً عن طريق تكوين بارا تولودين أمين، أو في حالة تكوين المركبين الناتجين عن طريق أحد المركبات المتوسطة العادية، فحينئذ كلاهما لا يمكن الحصول عليها بطريقة الاحلال المباشرة البسيطة.
 في محلول النشادر لا يتم الحصول على بارا تولودين المتوقع وجوده فقط بل نجد أيضاً المركب غير المتوقع الحصول عليه إطلاقاً وهو ميتاً تولودين أمين من بارا – كلوروتلوين بطريقة الإحلال البسيطة غير ممكن، ويكون تكوينه اما عن طريق يختلف تماماً عن طريق تكوين بارا تولودين أمين، أو في حالة تكوين المركبين الناتجين عن طريق أحد المركبات المتوسطة العادية، فحينئذ كلاهما لا يمكن الحصول عليها بطريقة الاحلال المباشرة البسيطة.")
189
التفاعل الذي يسير بطريقة غير متوقعة سواء كان سريعاً أم بطيئاً بالنسبة إلى غيره من تفاعلات المركبات ذات العلاقة التركيبية المشتركة تحت الشروط نفسها قد يشير إلى ميكانيكية تختلف عن التي تكون قد افترضت، فنجد أن المعدلات الملحوظة للتحلل المائي لمركبات الكلوروميثين في وجود القلويات القوية تحت شروط متقاربة تختلف بحسب التالي: CH3Cl >> CH2Cl2 >> CHCl3 >> CCl4 ومن الواضح أن هذا يقترح أن الكلوروفورم يعاني مداهمات بطرق تختلف عن المركبات الأخرى. مما سبق نستنتج أن درجة نجاح الميكانيكية المقترحة التي يمكن بها أن يصور ويرسم خط تفاعل لا يمكن اقتصارها على قدرة التفاعل على تعليل الحقائق المعروفة بل يجب الاهتمام بالوسيلة التي يتم بها تطبيق القليل نسبياً من الأسس والقواعد التوجيهية البسيطة والتي تمكن من إلقاء الضوء على الكثير من المعلومات المتفاوتة عن حالات الاتزان وطاقة الحركة وسرعات التفاعل وكذلك النشاط النسبي للتفاعل.

190
المراجع 1- الكيمياء الحركية: د. رضا محمد سعيد عبيد كلية العلوم جامعة الرياض (جامعة الملك سعود حالياً)، الطبعة ألأولى، 1974م. 2- S.Glasstone and D.Lewis, Elements of physical Chemistry, The macmillan Prees. Ltd. 2 nd Ed (1961). 3- C.Castellan , Physical Chemistry , Addison –Wesley Publishing Company , 2nd Ed (1971). 4- S. Glasstone, physical Chemistry, S. G. Wasani for the Macmillan Co. of India Limited , 2nd Ed (1977). 5- M.Barrow, Physical Chemistry , Untrenational Student Edition 3 rd Ed., (1961). 6- J.Bares, C.Cerny, V.Fried and J.Pick, Collection of Problems in Physical Chemistry, Pergamon Press New Delhi (1974). رسالة الماجستير للمعيدة سناء طاهر عرب تحت إشراف د.ليلى عبد الرحمن الحسن محرم (1403).
، الطبعة ألأولى، 1974م. 2- S.Glasstone and D.Lewis, Elements of physical Chemistry, The macmillan Prees. Ltd. 2 nd Ed (1961). 3- C.Castellan , Physical Chemistry , Addison –Wesley Publishing Company , 2nd Ed (1971). 4- S. Glasstone, physical Chemistry, S. G. Wasani for the Macmillan Co. of India Limited , 2nd Ed (1977). 5- M.Barrow, Physical Chemistry , Untrenational Student Edition 3 rd Ed., (1961). 6- J.Bares, C.Cerny, V.Fried and J.Pick, Collection of Problems in Physical Chemistry, Pergamon Press New Delhi (1974). رسالة الماجستير للمعيدة سناء طاهر عرب تحت إشراف د.ليلى عبد الرحمن الحسن محرم (1403).")
عروض تقديميّة مشابهة
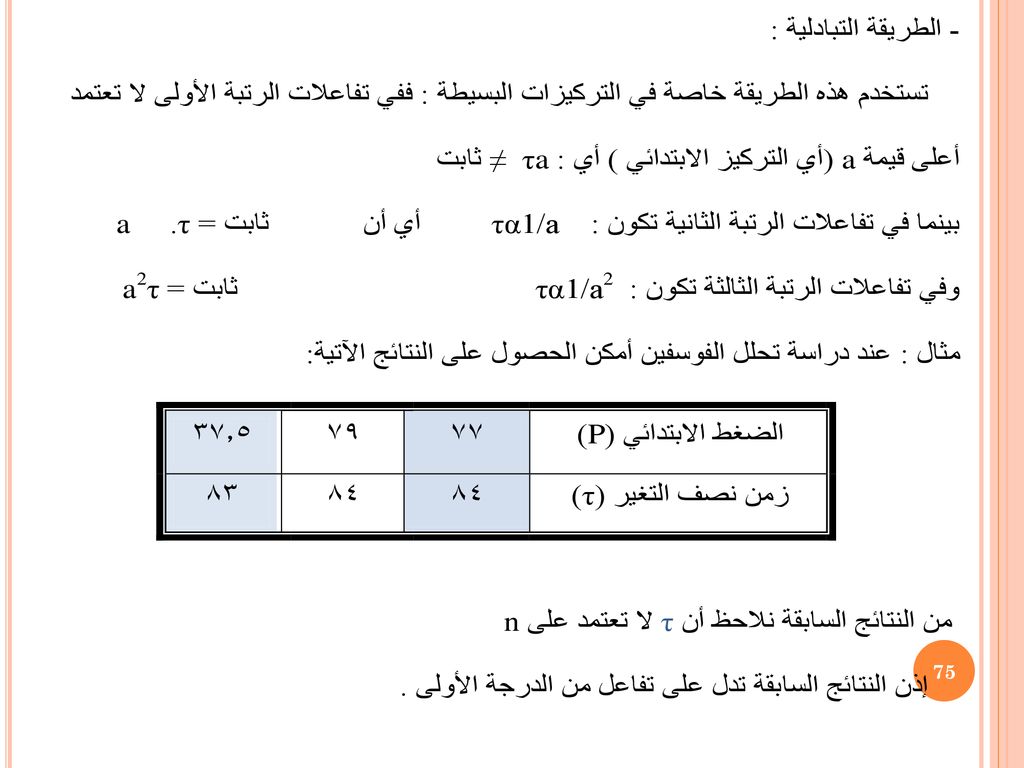




 استعداد المجتمع المحلّي *>")


